- Home
- Medicamentos
- Medicamentos Especiais
- Tafinlar 50Mg, Embalagem Conte...
- Bula de Tafinlar 50Mg, Embalagem Conte...
para o que é indicado e para que serve?
Para que serve Tafinlar é indicado para o tratamento de melanoma, um tipo de câncer de pele, quando este se espalhou pelo corpo e não pode ser removido por cirurgia e para pacientes com mutação no gene BRAF V600E.Continue lendo...
ofertas de Tafinlar 50Mg, Embalagem Contendo 120 Cápsulas

ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Tafinlar é indicado para o tratamento de melanoma, um tipo de câncer de pele, quando este se espalhou pelo corpo e não pode ser removido por cirurgia e para pacientes com mutação no gene BRAF V600E.
Como o Tafinlar funciona?
O dabrafenibe, composto presente em Tafinlar, é um inibidor de alguns tipos de enzimas BRAF. As mutações oncogênicas em BRAF levam a estimulação do crescimento das células tumorais e têm sido identificadas em uma alta frequência em cânceres específicos, incluindo aproximadamente 50% dos melanomas. O dabrafenibe inibe o crescimento celular de melanoma mutante BRAF V600 in vitro e in vivo.
Contraindicação
Tafinlar é contraindicado para pacientes com hipersensibilidade (alergia) a qualquer componente da formulação.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Informe imediatamente seu médico em caso de suspeita de gravidez.
Como usar
O tratamento com Tafinlar deve ser iniciado por um médico com experiência em terapias antineoplásicas.
Tafinlar só deve ser utilizado para o tratamento de melanomas com mutação no gene BRAF. Desta forma, antes do início do tratamento com Tafinlar, seu médico irá solicitar exames para determinar se você possui mutação BRAF V600.
Modo de uso
Uso oral.
Sempre tome Tafinlar exatamente conforme as instruções do seu médico. As cápsulas devem ser tomadas inteiras, com o auxílio de água.
Tafinlar deve ser tomado pelo menos uma hora antes, ou duas horas após as refeições, deixando um intervalo de aproximadamente 12 horas entre as doses. Tafinlar deve ser tomado em horários semelhantes todos os dias.
Posologia
Adultos
A dose recomendada é de 150mg duas vezes ao dia (de 12 em 12 horas), correspondendo a uma dose diária total de 300mg.
Seu médico irá determinar, com base na progressão da doença e no desenvolvimento de toxicidade, a duração do tratamento.
Ajustes da dose
De acordo com os sinais e sintomas desenvolvidos, como, por exemplo, febre (temperatura corpórea ? 38,5°C) e uveíte (inflamação nos olhos), o médico poderá recomendar interrupções no tratamento, redução de dose ou descontinuação do tratamento com Tafinlar.
Posologia para Populações Especiais
Crianças e adolescentes (abaixo de 18 anos)
A eficácia e segurança de Tafinlar não foram estabelecidas em crianças e adolescentes (<18 anos).
Tafinlar não é recomendado para essa faixa etária.
Idosos (65 anos de idade ou acima)
Nenhum ajuste de dose é necessário para pacientes acima de 65 anos.
Insuficiência renal (nos rins)
Nenhum ajuste de dose é exigido para pacientes com comprometimento renal leve a moderado. Tafinlar deve ser usado com cautela em pacientes com insuficiência renal grave.
Insuficiência hepática (no fígado)
Nenhum ajuste de dose é exigido para pacientes com comprometimento hepático leve. Tafinlar deve ser usado com cautela em pacientes com comprometimento hepático moderado à grave.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento de seu médico.
Este medicamento não deve ser aberto ou mastigado.
O que devo fazer quando eu me esquecer de usar o Tafinlar?
Se você se esquecer de tomar o medicamento e se lembrar com até seis horas de atraso, tome assim que se lembrar.
Se você se esquecer de tomar o medicamento e se lembrar com mais de seis horas, não use uma dose duplicada para repor a dose esquecida. Apenas siga com o tratamento, tomando normalmente a medicação no dia seguinte.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Converse atentamente com seu médico antes de usar Tafinlar se você:
- Tem algum problema no fígado. Seu médico poderá solicitar exames de sangue para avaliar a função do seu fígado durante seu tratamento com Tafinlar;
- Tem algum problema nos rins.
Algumas pessoas em tratamento com Tafinlar podem desenvolver outras condições, que podem ser graves.
Febre
Foi relatada febre em alguns pacientes fazendo uso de Tafinlar. Em uma minoria de casos, a febre foi acompanhada por rigores graves, desidratação, hipotensão (pressão sanguínea baixa) que em alguns casos levaram à insuficiência renal (dos rins) aguda. O seu médico irá solicitar exames de sangue para avaliar a função dos rins durante e após eventos de febre. Ele poderá também recomendar a interrupção do tratamento e/ou a redução da dose de Tafinlar, ou o uso de outros medicamentos concomitantes e fará um acompanhamento para avaliar a presença de sintomas ou sinais de infecção. Informe seu médico imediatamente se você apresentar febre durante o tratamento com Tafinlar.
Carcinoma de células escamosas cutâneo (cuSCC)
Casos de carcinoma de células escamosas cutâneo, um tipo de tumor em que a superfície da pele fica crostosa e descamativa, têm sido relatados em pacientes tratados com Tafinlar. Normalmente as lesões permanecem locais, podem ser removidas com cirurgia e os pacientes podem continuar o tratamento. Exame de pele deve ser realizado antes do início de Tafinlar e durante o tratamento com Tafinlar, a cada 2 meses durante a terapia e a cada 2 ou 3 meses por até 6 meses após a descontinuação de Tafinlar. O seu médico irá recomendar o melhor tratamento para você e Tafinlar deve ser continuado sem qualquer ajuste de dose. Você deve informar imediatamente ao seu médico se desenvolver novas lesões.
Novo melanoma primário
Novo melanoma primário tem sido relatado em pacientes tratados com Tafinlar. Em estudos clínicos em melanoma, estes foram identificados dentro dos 5 primeiros meses de terapia e não necessitaram de outra modificação no tratamento além da excisão. O monitoramento para lesões de pele deve ocorrer como descrito para carcinoma de células escamosas cutâneo.
Malignidade não cutânea
Pacientes tratados com Tafinlar podem ter um risco aumentado de apresentar outros tipos de câncer que não os de pele. Seu médico fará o monitoramento de seus sintomas e avaliará os benefícios e riscos de continuar o tratamento com Tafinlar. Ele poderá recomendar o monitoramento por até 6 meses após a interrupção do tratamento ou até o início de outra terapia antineoplásica.
Pancreatite (inflamação no pâncreas)
Em estudos clínicos em melanoma, pancreatite foi relatada em <1 % dos indivíduos tratados com Tafinlar.
Um dos eventos ocorreu no primeiro dia de dose e recorreu após reintrodução a uma dose reduzida. Caso apresente dores abdominais não explicáveis, consulte seu médico. Ele irá monitorar seus sintomas caso apresente pancreatite durante o tratamento com Tafinlar e após o reinício deste.
Reações oftalmológicas (nos olhos)
Reações oftalmológicas, incluindo uveíte e irites, inflamações na região dos olhos, têm sido relatadas. Durante o tratamento, seu médico irá monitorá-lo para a presença de sinais ou sintomas visuais, como alterações na visão, fotofobia (sensibilidade à luz) e irritação e dor nos olhos.
Reações Adversas
Como todo medicamento, Tafinlar pode provocar efeitos indesejáveis.
Os dados de segurança de Tafinlar foram obtidos a partir de estudos clínicos de dabrafenibe em monoterapia em pacientes com melanoma metastático ou irressecável com mutação de BRAF V600.
Reações muito comuns (ocorrem em mais de 10% dos pacientes que utilizam este medicamento):
Papiloma (verrugas que aparecem na superfície da pele), diminuição do apetite, dor de cabeça, tosse, enjoo, vômitos, diarreia, reações na pele como erupções e espessamento externo (rash cutâneo, hiperqueratose), queda de cabelo (alopecia), palmas das mãos ou solas dos pés adormecidas, inchadas, doloridas ou avermelhadas; dores nas articulações (artralgia), dores musculares (mialgia), dor nas extremidades, cansaço e fraqueza ao acordar (astenia), calafrios, cansaço, febre.
Reações comuns (ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento):
Acrocórdon (protuberâncias na pele), carcinoma de células escamosas cutâneo (SCC), protuberâncias pardas/marrons (queratose seborreica), diminuição nos níveis de fósforo no sangue (hipofosfatemia), prisão de ventre (constipação), efeitos de pele, incluindo manchas ásperas e descamativas, espessamento amarelado/amarronzado da pele, marcas na pele, pele seca e vermelhidão na pele (queratose actínica, lesão cutânea, pele seca, eritema, coceira), doença semelhante à gripe, hiperglicemia (aumento no nível de açúcar no sangue), nasofaringite (inflamação no nariz e garganta).
Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento):
Novo melanoma primário, reações alérgicas (hipersensibilidade), inflamações nos olhos (uveíte), inflamação no pâncreas (pancreatite), insuficiência renal (nos rins), insuficiência renal aguda, prolongamento do intervalo QT, paniculite (inflamação da camada gordurosa embaixo da pele).
Reações raras (ocorrem entre 0,1% e 0,01% dos pacientes que utilizam este medicamento):
Nefrite tubulointersticial (uma inflamação nos rins).
Avise seu médico imediatamente se você apresentar um ou mais desses sintomas. Eles podem persistir mesmo depois que você interromper o uso de Tafinlar.
Se um ou mais dos sintomas listados nesta bula se agravar ou se você observar algum sintoma que não tenha sido relacionado aqui, informe seu médico ou farmacêutico.
Atenção: este produto é um medicamento novo, e embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
População Especial
Crianças e adolescentes
Tafinlar não é recomendado para crianças e adolescentes. Os efeitos de Tafinlar em menores de 18 anos ainda não são conhecidos.
Insuficiência hepática (no fígado)
Insuficiência hepática leve não teve efeito significativo sobre Tafinlar. Não existem dados disponíveis em indivíduos com insuficiência hepática moderada a grave.
Insuficiência renal (nos rins)
Insuficiência renal leve ou moderada não teve efeito significativo sobre Tafinlar. Não existem dados disponíveis em indivíduos com insuficiência renal grave.
Idade
Baseado em dados de estudos, a idade não tem nenhum efeito significativo sobre Tafinlar.
Peso corporal e Sexo
Baseado em dados de estudos, sexo e peso corporal mostraram ter efeito sobre Tafinlar. No entanto, eles não foram considerados clinicamente relevantes.
Raça/Etnia
A análise dos dados dos estudos com Tafinlar não mostrou diferenças significativas na resposta de dabrafenibe entre pacientes asiáticos e caucasianos (brancos). Nenhum ajuste de dose é necessário em pacientes asiáticos.
Não há dados suficientes para avaliar o potencial efeito da raça sobre Tafinlar.
Gravidez
Tafinlar pode causar danos ao feto quando administrado a uma mulher grávida. Estudos em animais têm demonstrado que Tafinlar pode ser perigoso ao feto em desenvolvimento. Por isso, você deve evitar ficar grávida enquanto usar Tafinlar. Para isso, utilize um método contraceptivo (uma forma de evitar a gravidez) confiável durante o tratamento com Tafinlar e por 28 dias após a descontinuação do tratamento. Tafinlar pode diminuir a eficácia de contraceptivos hormonais e um método alternativo de contracepção, tais como preservativos, deve ser usado.
Informe seu médico imediatamente se você ficar grávida durante o tratamento com Tafinlar.
Amamentação
Não se recomenda amamentar durante o tratamento com Tafinlar. Não há dados do efeito de Tafinlar no lactente, e nem na produção de leite. Caso esteja amamentando ou planejando amamentar, informe seu médico.
Ele irá decidir se você deve amamentar ou seguir com o tratamento.
Infertilidade
Não há dados em humanos. Efeitos adversos nos órgãos reprodutivos masculinos têm sido observados em animais. Homens fazendo tratamento com Tafinlar podem apresentar uma redução na contagem de espermatozoides. A contagem de espermatozoides pode não retornar aos níveis normais após a interrupção do tratamento.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista. Informe imediatamente seu médico em caso de suspeita de gravidez.
Composição
Cada cápsula dura de 50mg de Tafinlar contém:
Dabrafenibe 50mg (equivalente a 59,25mg de mesilato de dabrafenibe).
Excipientes: celulose microcristalina, estearato de magnésio, dióxido de silício coloidal, óxido de ferro vermelho, dióxido de titânio, hipromelose, óxido de ferro preto, goma laca, álcool N-butil, álcool isopropílico, propilenoglicol e hidróxido de amônio.
Cada cápsula dura de 75mg de Tafinlar contém:
Dabrafenibe 75mg (equivalente a 88,88mg de mesilato de dabrafenibe).
Excipientes: celulose microcristalina, estearato de magnésio, dióxido de silício coloidal, óxido de ferro vermelho, dióxido de titânio, hipromelose, óxido de ferro preto, goma laca, álcool N-butil, álcool isopropílico, propilenoglicol e hidróxido de amônio.
Superdosagem
Sinais e Sintomas
Não existe nenhum antídoto específico para superdosagem com Tafinlar. Em caso de superdosagem, o tratamento com Tafinlar deve ser suspenso até a resolução dos sintomas. Procedimentos adicionais devem ser realizados conforme indicação de seu médico.
Tratamento
Se acidentalmente você tomar medicamento demais, deve falar com seu médico ou farmacêutico ou entrar em contato com o departamento de emergência do hospital mais próximo para obter instruções.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Efeitos de outros fármacos neste medicamento
Baseado em estudos in vitro, dabrafenibe mostrou ser metabolizado principalmente pelo citocromo P450 (CYP) CYP2C8 e CYP3A4. Dados farmacocinéticos demonstraram um aumento na Cmáx (33%) de dabrafenibe quando em doses repetidas e na AUC (71%) com a coadministração de cetoconazol (inibidor de CYP3A4); e aumentos de 82% e 68% nas AUC de hidroxi- e desmetil-dabrafenibe, respectivamente. Um decréscimo na AUC foi observado para carboxi-dabrafenibe (decréscimo de 16%). A coadministração de dabrafenibe e genfibrozila (um inibidor de CYP2C8) resultou em um aumento da AUC de dabrafenibe em doses repetidas (47%) e nenhuma alteração significativa nas concentrações dos metabólitos. Dados de farmacocinética em doses repetidas deste medicamento apresentaram uma diminuição na Cmáx (diminuição de 27%) e AUC (diminuição de 34%) mediante coadministração com rifampicina (indutor CYP3A4/ CYP2C8). Nenhuma alteração relevante na AUC fora verificada para hidroxi-dabrafenibe, e teve um aumento de 73% na AUC para carboxi-dabrafenibe e uma diminuição de 30% a AUC para desmetil-dabrafenibe.
Medicamentos que são forte inibidores ou indutores de CYP2C8 ou CYP3A4 são susceptíveis de aumentar ou diminuir, respectivamente, as concentrações de dabrafenibe.
Agentes alternativos devem ser considerados durante a administração com este medicamento, quando possível. Tenha cuidado com inibidores fortes (por exemplo, cetoconazol, nefazodona, claritromicina, ritonavir, genfibrozila) ou indutores (por exemplo, rifampicina, fenitoína, carbamazepina, fenobarbital, Erva de São João) de CYP2C8 ou CYP3A4 quando coadministrados com este medicamento.
Fármacos que afetam o pH gástrico
A administração concomitante de doses repetidas deste medicamento 150mg duas vezes ao dia e o agente de elevação de pH, rabeprazol 40mg uma vez ao dia, resultou em um aumento na AUC de 3% e uma diminuição de 12% na Cmáx. Essas alterações na AUC e Cmáx para este medicamento não são consideradas clinicamente significativas. Medicamentos que alteram o pH do trato gastrointestinal superior (por exemplo, inibidores de bomba de prótons, antagonistas de receptores H2 e antiácidos) não são esperados que diminuam a biodisponibilidade do dabrafenibe.
Efeito deste medicamento em outros fármacos
O dabrafenibe induz o metabolismo mediado por CYP3A4 e CYP2C9 e pode induzir outras enzimas incluindo CYP2B6, CYP2C8, CYP2C19 e UDP glucuronosiltransferase (UGT). O dabrafenibe também pode induzir transportadores [por exemplo, glicoproteína P (P-gp)].
Em um estudo clínico com 12 pacientes usando uma dose única de midazolam, um substrato da CYP3A4, a Cmáx e AUC tiveram diminuição em 61% e 74%, respectivamente com a coadministração de doses repetidas deste medicamento duas vezes ao dia. Em um estudo separado com 14 pacientes, doses repetidas deste medicamento diminuíram a AUC da S-varfarina em dose única (um substrato da CYP2C9) e da R-varfarina (um substrato de CYP3A4/CYP1A2) em 37% e 33%, respectivamente, com um pequeno aumento da Cmáx (18 e 19% respectivamente).
A coadministração deste medicamento e medicamentos que são afetados pela indução da CYP3A4 ou CYP2C9, tais como contraceptivos hormonais, varfarina, ou dexametasona podem resultar em concentrações diminuídas ou perda de eficácia (ver Gravidez e Lactação, em Advertências e Precauções). Se a coadministração destes medicamentos for necessária, monitorar os indivíduos para perda de eficácia ou considerar a substituição desses medicamentos.
Ação da Substância
Resultados de Eficácia
Estudos clínicos
A eficácia deste medicamento no tratamento de pacientes adultos com melanoma irressecável ou metastático positivo para mutação BRAF V600 tem sido avaliada em 3 estudos (BRF113683 [BREAK-3], BRF113929 [BREAK-MB], e BRF113710 [BREAK-2]) incluindo pacientes com BRAF V600E e/ou mutações do V600K.
Pacientes não tratados previamente
A segurança e eficácia deste medicamento foram avaliadas em um estudo fase III, randomizado, aberto, [BREAK-3] comparando este medicamento a dacarbazina (DTIC) em pacientes com melanoma positivo para mutação BRAF V600E avançado (irressecável Estágio III) ou metastático (Estágio IV) não tratados previamente. A triagem incluiu teste central da mutação de BRAF V600E usando um ensaio de mutação de BRAF realizado na amostra do tumor mais recente disponível.
O estudo incluiu 250 pacientes randomizados 3:1 para receber ou este medicamento 150 mg duas vezes ao dia ou DTIC intravenoso 1000 mg/m2 a cada 3 semanas. O objetivo primário para este estudo era avaliar a eficácia deste medicamento comparado ao DTIC com relação à sobrevida livre de progressão (PFS) para pacientes com melanoma metastático com mutação positiva para BRAF V600E. Aos pacientes no braço do DTIC foi permitido receber este medicamento independente após confirmação radiográfica de progressão inicial. As características do período basal foram balanceadas entre os grupos de tratamento. Sessenta por cento dos pacientes eram homens e 99,6% eram caucasianos, a idade média era de 52 anos com 21% dos pacientes sendo ! 65 anos, 98,4% tinham condição de ECOG (Eastern Cooperative Oncology Group) de 0 ou 1, e 97% dos pacientes tinham doença metastática.
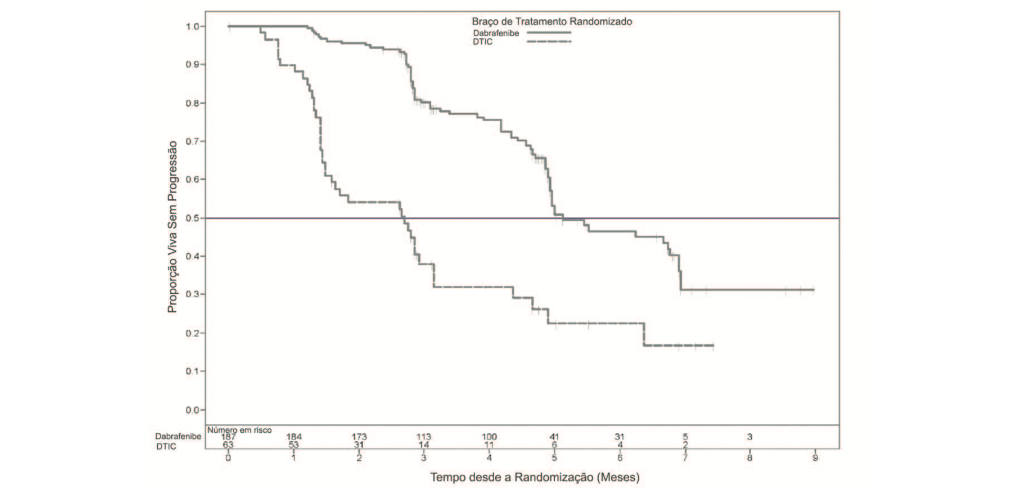
A análise primária foi baseada em 118 eventos no momento de corte de dados. A avaliação do investigador para os dados de eficácia estão resumidos na Tabela 1 e Figura 1.
Tabela 1: Dados de eficácia por avaliação do investigador para o Estudo BREAK-3
|
População com Intenção de Tratar | ||
|
Endpoints / Avaliação |
este medicamento N=187 |
DTIC N=63 |
|
Sobrevida livre de progressão (avaliação do investigador) | ||
|
Mediana, meses (95 % IC) HR (95 % IC) |
5,1 (4,9, 6,9) 2,7 (1,5, 3,2) 0,30 (0,18, 0,51) | |
|
Sobrevida Globala | ||
|
% até 6 meses (95 % IC) HR (95 % IC) |
87 (79,2, 91,9) 79 (59,7, 89,5) 0,61 (0,25, 1,48) | |
|
Resposta Globalb | ||
|
% (95 % IC)c |
53 (45,5, 60,3) |
19 (10,2, 30,9) |
|
Duração da resposta | ||
|
Mediana, meses (95 % IC) |
N=99 |
N=12 |
| Abreviações: IC: intervalo de confiança; DTIC: dacarbazina; HR: Hazard ratio; NR:não alcançado a Estimado a partir de curvas de Kaplan-Meier de 6 meses; Com a mediana de tempo de seguimento de 4,9 meses (alcance = 0 a 9,9 meses) e 30 mortes, dados de sobrevida global ainda não estão maduros e mediana de sobrevida global não foi atingida por nenhum dos braços. Indivíduos são resumidos pelo tratamento randomizado; as estimativas incluem dados da fase de cruzamento para indivíduos randomizados para DTIC e, portanto, reflete qualquer benefício de segunda linha deste medicamento. b Definida como resposta completa+resposta parcial. c Resposta confirmada. | ||
Vinte e oito indivíduos (44 %) randomizados para DTIC cruzaram para este medicamento seguindo a progressão da doença verificada de forma independente. O tempo mediano neste medicamento após o cruzamento foi de 2,8 meses e a taxa de resposta global (ORR) não confirmada foi de 46%.
Figura 01: Avaliação Kaplan-Meier do investigador da sobrevida livre de progressão– pacientes não tratados previamente (população ITT)
Pacientes com metástases cerebrais
BREAK-MB foi um estudo multicêntrico, aberto, de duas coortes, de Fase II desenhado para avaliar a resposta intracranial deste medicamento em indivíduos com confirmação histológica (Estágio IV) de melanoma com mutação positiva BRAF (V600E ou V600K) metastático para o cérebro. Os indivíduos foram incluídos na Coorte A (indivíduos sem tratamento local prévio para metástases cerebrais) ou Coorte B (indivíduos que receberam tratamento local prévio para metástases cerebrais). Os resultados estão resumidos na Tabela 02.
Tabela 2: Dados de eficácia em pacientes com metástases no cérebro (Estudo BREAK-MB)
|
Toda População de Indivíduos Tratados | ||||
|
BRAF V600E (Primário) |
BRAF V600K | |||
|
Endpoints / Avaliação |
Coorte A N=74 |
Coorte B N=65 |
Coorte A N=15 |
Coorte B N=18 |
|
Taxa de resposta intracraniana global, % (95 % IC)a | ||||
|
39% (28,0, 51,2) P < 0,001b |
31% (19,9, 43,4) P < 0,001b |
7% (0,2, 31,9) |
22% (6,4, 47,6) | |
|
Duração da resposta intracraniana, mediana, meses (95% IC) | ||||
|
N=29 |
N=20 |
N=1 |
N=4 | |
|
Resposta Global, % (95% IC)a | ||||
|
38% (26,8, 49,9) |
31% (19,9, 43,4) |
0 (0, 21,8) |
28% (9,7, 53,5) | |
|
Duração da resposta, mediana, meses (95% IC) | ||||
|
N=28 |
N=20 |
NA |
N=5 | |
|
Sobrevida livre de progressão, mediana, meses (95% IC) | ||||
|
3,7 (3,6, 5,0) |
3,8 (3,6, 5,5) |
1,9 (0,7, 3,7) |
3,6 (1,8, 5,2) | |
|
Sobrevida Global, mediana, meses (95% IC) | ||||
|
Mediana, meses |
7,6 (5,9, NR) |
7,2 (5,9, NR) |
3,7 (1,6, 5,2) |
5,0 (3,5, NR) |
| Abreviações: IC: intervalo de confiança; NR: não alcançado; NA: não aplicável a – Resposta confirmada. b – Este estudo foi desenhado para apoiar ou rejeitar a hipótese nula de OIRR% ? 10 (com base nos resultados históricos) em favor da hipótese alternativa de OIRR ? 30% em indivíduos BRAF V600E positivos. | ||||
Pacientes que não foram tratados previamente ou falharam em pelo menos uma terapia sistêmica prévia
BRF113710 (BREAK-2) foi um estudo multicêntrico, global, aberto, braço único, de Fase II que incluiu 92 indivíduos de pesquisa com melanoma metastático confirmado histologicamente (Estágio IV) com melanoma positivo para mutação BRAF V 600E ou V600K confirmada. Os indivíduos eram virgens de tratamento (n=15) ou receberam tratamento prévio (n=77) na presença de metástases (por exemplo, quimioterapia, imunoterapia, terapia alvo prévia, etc.).
O investigador avaliou a taxa de resposta confirmada na eficácia primária em uma população de pacientes com melanoma metastático BRAF V600E (n=76) foi de 59 % (95% IC: 48,2; 70,3) incluindo 7% de resposta completa. A mediana de PFS foi de 6,3 meses (95% IC: 4,6; 7,7) e a duração mediana da resposta foi de 5,2 meses (95 % IC: 3,9, não calculado). A terapia sistêmica prévia não pareceu afetar significativamente a resposta. O investigador avaliou a taxa de resposta confirmada na eficácia secundária em uma população de pacientes com melanoma metastático com mutação positiva BRAF V600K (n=16) foi de 13% (95% IC: 0,0; 28,7) com duração mediana da resposta de 5,3 meses (95 % IC: 3,7; 6,8). Não houve resposta completa na população de pacientes V600K.
Características Farmacológicas
Propriedades Farmacodinâmicas
Efeitos Farmacodinâmicos
O dabrafenibe demonstrou supressão de um biomarcador farmacodinâmico à jusante (ERK fosforilado) em linhagens celulares de melanoma mutante BRAF V600, in vitro e em modelos animais.
Em indivíduos com melanoma mutante BRAF V600, a administração deste medicamento resultou em inibição de ERK fosforilado do tumor em relação ao período basal.
Eletrofisiologia cardíaca: O efeito potencial do dabrafenibe no prolongamento QT foi avaliado em um estudo QT com múltiplas doses. Uma dose supraterapêutica de 300 mg de dabrafenibe duas vezes ao dia foi administrada em 32 indivíduos com tumores com mutação BRAF- V600 positiva. Nenhum evento clinicamente relevante de dabrafenibe ou seus metabólitos no intervalo QTc foi observado.
Propriedades Farmacocinéticas
Absorção
O dabrafenibe é absorvido oralmente com tempo mediano para alcançar o pico de concentração plasmática de 2 horas após a dose. A biodisponibilidade absoluta média de dabrafenibe oral é de 95 % (90% IC: 81;110). A exposição ao dabrafenibe (Cmáx e AUC) aumentou de uma forma proporcional à dose entre 12 e 300 mg seguindo a administração de dose única, mas o aumento foi menor que proporcional a dose após repetir a dose duas vezes ao dia. Houve uma diminuição na exposição observada com a repetição da dose, provavelmente devido à indução de seu próprio metabolismo. A razão de acumulação média de AUC dia 18/Dia 1 foi 0,73. Após administração de 150 mg duas vezes ao dia, a média geométrica Cmáx, AUC(0-t) e a concentração pré-dose (Ct) foram 1478 ng/mL, 4341 ng*hr/mL e 26 ng/mL, respectivamente.
A administração deste medicamento com comida reduziu a biodisponibilidade (Cmáx e AUC diminuíram para 51% e 31% respectivamente) e retardou a absorção de dabrafenibe em cápsulas, quando comparado ao estado de jejum.
Distribuição
O dabrafenibe liga-se a proteína plasmática humana e é 99,7% ligado. A distribuição do volume em estado de equilíbrio após administração intravenosa de microdose é 46 L.
Biotransformação/Metabolismo
O metabolismo de dabrafenibe é mediado principalmente por CYP2C8 e CYP3A4 para formar hidroxi-dabrafenibe, o qual é posteriormente oxidado via CYP3A4 para formar carboxi-dabrafenibe. Carboxi-dabrafenibe pode ser descarboxilado via um processo não enzimático para formar desmetil-dabrafenibe. Carboxi-dabrafenibe é excretado na bile e urina. Desmetil-dabrafenibe também pode ser formado no intestino e reabsorvido. Desmetil-dabrafenibe é metabolizado pelo CYP3A4 a metabólitos oxidativos. A meia-vida terminal de hidroxi-dabrafenibe corresponde àquela do parental com meia-vida de 10 horas enquanto que os metabólitos carboxi- e desmetil- exibiram meia-vidas mais longas (21-22 horas). As médias da razão de AUC parental do metabólito após a administração de dose repetida foram 0,9, 11 e 0,7 para hidroxi-, carboxi- e desmetil-dabrafenibe, respectivamente. Com base na exposição, potência relativa e propriedades farmacocinéticas, tanto para hidroxi- e desmetil-dabrafenibe são passíveis de contribuir para a atividade clínica deste medicamento, enquanto que a atividade do carboxi- dabrafenibe não é passível de ser significativa.
Eliminação
A meia-vida terminal seguida de microdose IV é de 2,6 horas. A meia-vida terminal de dabrafenibe é de 8 horas devido a uma fase terminal prolongada após a administração oral. O clearance plasmático IV é 12 L/hr.
A excreção fecal é a rota principal de eliminação após a administração oral, representando 71 % da dose radioativa enquanto a excreção urinária representa 23 % da radioatividade.
Populações Especiais
Insuficiência Hepática
A farmacocinética de dabrafenibe foi caracterizada em 65 pacientes com insuficiência hepática leve (com base na classificação do Instituto Nacional do Câncer [NCI]) incluídos em estudos clínicos, utilizando uma análise da população. O clearance do dabrafenibe oral não foi significativamente diferente entre estes indivíduos e indivíduos com função hepática normal (4% de diferença). Além disso, insuficiência hepática leve não teve um efeito significante na concentração plasmática do metabólito de dabrafenibe. Não há dados disponíveis em pacientes com insuficiência hepática moderada a grave (ver Posologia e Modo de Usar).
Insuficiência Renal
A farmacocinética de dabrafenibe foi caracterizada em 233 pacientes com insuficiência renal leve (GFR 60-89 mL/min/1,73 m2) e em 30 pacientes com comprometimento renal moderado (GFR 30-59 mL/min/1,73 m2) incluídos em estudos clínicos utilizando análise de população. O efeito da insuficiência renal leve ou moderada no clearance oral de dabrafenibe foi pequeno < 6%, para ambas as categorias) e não foi clinicamente relevante. Além disso, insuficiência renal leve ou moderada não teve efeito significativo nas concentrações plasmáticas de hidroxi-, carboxi- e desmetil-dabrafenibe. Não existem dados disponíveis em indivíduos com insuficiência renal grave (ver Posologia e Modo de Usar).
Idade
Baseado na análise farmacocinética de população, a idade não tem nenhum efeito significativo na farmacocinética de dabrafenibe. Idade superior a 75 anos foi um preditor significativo das concentrações plasmáticas de carboxi- e desmetil-dabrafenibe com uma exposição 40% maior em indivíduos ? 75 anos de idade, em relação aos indivíduos <75 anos de idade.
Peso corporal e Sexo
Baseados na análise farmacocinética de população, sexo e peso corporal mostraram influenciar o clearance oral de dabrafenibe; o peso também impactou a distribuição do volume oral e o clearance distributivo. Estas diferenças farmacocinéticas não foram consideradas clinicamente relevantes.
Raça/Etnia
A análise farmacocinética da população não mostrou diferenças significativas na farmacocinética de dabrafenibe entre pacientes asiáticos e caucasianos. Nenhum ajuste de dose é necessário em pacientes asiáticos.
Não há dados suficientes para avaliar o potencial efeito da raça sobre a farmacocinética de dabrafenibe.
Avaliação in vitro do potencial de interações medicamentosas
Efeito deste medicamento em outros fármacos
Em hepatócitos humanos, dabrafenibe produziu aumento concentração-dependente nos níveis de RNAm de CYP2B6 e CYP3A4 em até 32 vezes os níveis de controle.
Embora dabrafenibe e seus metabólitos hidroxi-dabrafenibe, carboxi-dabrafenibe e desmetil-dabrafenibe, sejam inibidores do polipeptídio transportador de ânions orgânicos humanos (OATP) 1B1, OATP1B3, transportador de ânion orgânico (OAT)1 e AOT3 e devido ao fato que dabrafenibe e seu metabólito desmetil demonstraram serem inibidores do transportador de cátions orgânicos 2 (OCT2) in vitro, o risco de interação medicamentosa é mínimo com base na exposição clínica.
O dabrafenibe e desmetil-dabrafenibe demonstraram serem inibidores moderados da BCRP; porém, com base na exposição clínica, o risco de interação medicamentosa é mínimo.
Nem dabrafenibe ou seus 3 metabólitos demonstraram ser inibidores da Pgp in vitro.
Efeito de outros fármacos neste medicamento:
Resultados de estudos in vitro indicam que CYP2C8 e CYP3A4 são as principais enzimas CYP envolvidas no metabolismo oxidativo do dabrafenibe, enquanto hidroxi-dabrafenibe e desmetil-dabrafenibe são substratos do CYP3A4. Portanto, inibidores ou indutores dessas enzimas têm o potencial de afetar a farmacocinética do dabrafenibe ou de seus metabólitos (ver Interações Medicamentosas).
O dabrafenibe é um substrato da Pgp humana e da Proteína de Resistência ao Câncer de Mama (BCRP)1 in vitro. Entretanto, estes transportadores tiveram um impacto mínimo na biodisponibilidade oral de dabrafenibe e na eliminação, e o risco de interação medicamentosa é mínimo.
Cuidados de Armazenamento
Mantenha o produto na embalagem original e em temperatura ambiente (entre 15°C e 30°C). Não remover o dessecante.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspectos físicos / Características organolépticas
Cápsulas 50mg
As cápsulas de 50mg são opacas, compostas por um corpo vermelho escuro e uma tampa vermelha escura. São gravadas com ‘GS TEW’ em uma das faces e ‘50mg’ na outra. O conteúdo da cápsula é um sólido branco a quase branco.
Cápsulas 75mg
As cápsulas de 75mg são opacas, compostas por um corpo rosa escuro e uma tampa rosa escura. São gravadas com ‘GS LHF’ em uma das faces e ‘75mg’ na outra. O conteúdo da cápsula é um sólido branco a quase branco.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
M.S: 1.0107.0319
Farm. Resp.:
Edinilson da Silva Oliveira
CRF-RJ Nº 18875
Fabricado por:
Glaxo Operations UK Limited
Priory Street, Ware, Hertfordshire, SG12 0DJ, Inglaterra
ou
GlaxoSmithKline Inc.
7333 Mississauga Road North, Mississauga, Ontário L5N 6L4 - Canadá
Embalado por:
Glaxo Wellcome S.A.
Avenida de Extremadura, 3-09400, Aranda de Duero (Burgos) - Espanha
Registrado e Importado por: GlaxoSmithKline Brasil Ltda.
Estrada dos Bandeirantes, 8464 – Rio de Janeiro – RJ
CNPJ: 33.247.743/0001-10
Venda sob prescrição médica.
informações complementares
| Fabricante |
| NOVARTIS |
| Princípio ativo |
| Mesilato De Dabrafenibe |
| Categoria do medicamento |
| Medicamentos Especiais |
TAFINLAR 50MG, EMBALAGEM CONTENDO 120 CÁPSULAS É UM MEDICAMENTO, NÃO USE SEM PRESCRIÇÃO MÉDICA E ORIENTAÇÃO DO FARMACÊUTICO. AO PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO.