- Home
- Medicamentos
- Medicamentos Especiais
- Tarceva 100Mg 30 Comprimidos
- Bula de Tarceva 100Mg 30 Comprimidos
para o que é indicado e para que serve?
Para que serve Câncer de pulmão de não pequenas células Tarceva é indicado para o tratamento de primeira linha e de manutenção de pacientes com câncer de pulmão do tipo não pequenas células (CPNPC), localmente avançado ou metastático, com mutações ativadoras de EGFR (receptor do fator de crescimento epidérmico).Continue lendo...
ofertas de Tarceva 100Mg 30 Comprimidos

ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Câncer de pulmão de não pequenas células
Tarceva é indicado para o tratamento de primeira linha e de manutenção de pacientes com câncer de pulmão do tipo não pequenas células (CPNPC), localmente avançado ou metastático, com mutações ativadoras de EGFR (receptor do fator de crescimento epidérmico).
No tratamento de manutenção, nenhum benefício clinicamente relevante foi demonstrado em pacientes com CPNPC sem mutação ativadora de EGFR.
Tarceva é indicado também para o tratamento de pacientes com câncer de pulmão do tipo não pequenas células (CPNPC) localmente avançado ou metastático, após a falha de pelo menos um esquema quimioterápico prévio.
Câncer de pâncreas
Tarceva, em combinação com gencitabina, é indicado para o tratamento de primeira linha de pacientes com câncer pancreático localmente avançado, inoperável ou metastático.
Peça ao seu médico para lhe explicar melhor sobre a sua doença.
Como Tarceva funciona?
Tarceva inibe a ação de uma enzima chamada tirosinoquinase presente em células normais e cancerosas.
Na célula cancerosa, Tarceva bloqueia a proliferação, podendo levá-la a morte, diminuindo, dessa forma, o tamanho do tumor.
Contraindicação
Tarceva está contraindicado a pacientes com hipersensibilidade severa a erlotinibe ou a qualquer componente da fórmula.
Como usar
Câncer de pulmão de não pequenas células
A dose diária recomendada de Tarceva é de 150 mg, por via oral, pelo menos uma hora antes ou duas horas depois da ingestão de alimentos.
Câncer de pâncreas
A dose diária recomendada de Tarceva é de 100 mg, por via oral, pelo menos uma hora antes ou duas horas depois da ingestão de alimentos, em combinação com gencitabina (vide as informações de gencitabina para a indicação de câncer de pâncreas).
O uso concomitante de medicamentos que utilizam a mesma via de metabolização hepática de Tarceva pode exigir ajuste da dose.
Quando for necessário ajuste da dose, recomenda-se reduzir em escalas de 50 mg.
Insuficiência hepática:
Se você tiver insuficiência hepática moderada, seu médico deve ter cautela ao lhe prescrever Tarceva e poderá reduzir a dose ou interromper o tratamento caso ocorram efeitos adversos graves, embora a exposição a erlotinibe tenha sido similar nesses pacientes. A segurança e a eficácia não foram estudadas em pacientes com insuficiência hepática grave.
Insuficiência renal:
A segurança e a eficácia de Tarceva não foram estudadas em pacientes com insuficiência renal.
Uso pediátrico:
A segurança e a eficácia de Tarceva não foram estudadas em pacientes com idade abaixo de 18 anos.
Fumantes:
O fumo de cigarros mostrou reduzir a exposição de erlotinibe. A dose máxima tolerada para fumantes ativos com câncer de pulmão de não pequenas células foi de 300 mg.
A eficácia e a segurança a longo prazo da dose superior à recomendada inicialmente não foram estabelecidas para pacientes que continuam fumando cigarros.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido ou mastigado.
O que devo fazer quando eu me esquecer de usar Tarceva?
Se você se esquecer de tomar uma ou mais doses de Tarceva, contate seu médico ou farmacêutico assim que possível. Não duplique a dose para compensar a dose esquecida.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Doença pulmonar intersticial:
Se você desenvolver quadro de novos sintomas pulmonares inexplicados ou progressivos, como dispneia (falta de ar), tosse e febre, procure seu médico, pois o tratamento com Tarceva deve ser interrompido e deve-se aguardar avaliação do seu médico.
Se você apresentar diagnóstico positivo para Doença Pulmonar Intersticial (DPI), Tarceva deve ser interrompido e iniciado tratamento apropriado, se necessário.
Diarreia, desidratação, desequilíbrio eletrolítico e insuficiência renal:
Caso você apresente diarreia grave ou persistente, náusea, anorexia ou vômitos associados à desidratação, procure seu médico, pois a terapia com Tarceva deve ser interrompida, e medidas apropriadas devem ser instituídas para tratar a desidratação (diminuição do potássio no sangue) e insuficiência renal secundária (incluindo óbitos).
Alguns relatos de falência renal foram secundários à desidratação severa causada por diarreia, vômito e / ou anorexia, enquanto outros foram associados à quimioterapia concomitante.
Em casos de diarreia grave ou persistente ou casos que levam à desidratação, particularmente em grupos de pacientes com fatores de risco agravantes (medicamentos concomitantes, sintomas ou outras condições predispostas, incluindo idade avançada), a terapia com Tarceva deve ser interrompida, e medidas apropriadas devem ser tomadas para hidratação intravenosa intensiva dos pacientes.
Além do mais, a função renal e os eletrólitos séricos, incluindo potássio, devem ser monitorados em pacientes com risco de desidratação.
Hepatite e insuficiência hepática:
Se você possui insuficiência hepática, testes periódicos de função do fígado devem ser considerados. A dosagem de Tarceva deve ser interrompida se ocorrerem mudanças graves na função hepática.
A segurança e a eficácia não foram estudadas em pacientes com disfunção hepática severa.
Perfurações gastrintestinais:
Pacientes tratados com Tarceva podem apresentar perfurações gastrintestinais, as quais foram observadas de forma rara (incluindo alguns casos fatais).
Se você estiver recebendo concomitantemente agentes antiangiogênicos (medicamentos utilizados para tratar câncer de pulmão), Tarceva, corticosteroides (prednisolona), anti-inflamatórios não esteroidais (AINEs) e / ou quimioterapia baseada em taxano (paclitaxel) ou se tiver histórico prévio de úlcera péptica ou doença diverticular (inflamação do intestino), você terá mais chances de ter perfurações gastrintestinais.
O tratamento com Tarceva deve ser permanentemente descontinuado se você desenvolver perfuração gastrintestinal.
Distúrbios bolhosos e esfoliativos da pele:
Foram relatadas condições bolhosas, vesiculares ou esfoliativas da pele, incluindo muito raramente casos sugestivos de síndrome de Stevens-Johnson / necrólise epidérmica tóxica, os quais, em alguns casos, foram fatais.
O tratamento com Tarceva deve ser interrompido ou descontinuado pelo seu médico se você apresentar bolhas, vesículas e esfoliações graves de pele.
Distúrbios oculares:
Casos muito raros de perfurações ou ulcerações da córnea foram relatados durante o uso de Tarceva.
Outros distúrbios oculares, incluindo crescimento anormal dos cílios, ceratoconjuntivite sicca ou ceratite, foram observados no tratamento com Tarceva, os quais também são fatores de risco para ulceração / perfuração da córnea.
O tratamento com Tarceva deve ser interrompido ou descontinuado pelo seu médico se você apresentar alterações oftalmológicas graves ou agravamento de distúrbios oculares, tais como dor nos olhos.
Insuficiência renal:
A segurança e a eficácia de Tarceva não foram estudadas em pacientes com insuficiência renal.
Uso pediátrico:
A segurança e a eficácia de Tarceva não foram estudadas em pacientes com idade abaixo de 18 anos.
Este medicamento não foi testado em pacientes com metástases cerebrais sintomáticas, e, portanto, sua eficácia é desconhecida nesse grupo de pacientes.
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas:
Não foram realizados estudos dos efeitos sobre a capacidade de dirigir veículos ou operar máquinas.
No entanto, erlotinibe não está associado com comprometimento da capacidade mental.
Gravidez e amamentação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Interações:
Tarceva pode reagir com outros medicamentos que você estiver tomando, além dos que estão citados a seguir. Informe ao seu médico se você fuma.
Os fumantes devem ser aconselhados a parar de fumar, pois o cigarro reduz a quantidade de erlotinibe no seu organismo em 50% – 60% e pode prejudicar o efeito de Tarceva.
Os pacientes não fumantes tiveram melhores resultados com Tarceva.
Reações Adversas
Experiência de estudos clínicos
A avaliação de segurança de Tarceva é baseada nos dados de mais de 1.500 pacientes tratados com, pelo menos, uma dose de 150 mg do medicamento em monoterapia e mais de 300 pacientes que receberam Tarceva 150 mg ou 100 mg em combinação com gencitabina.
A incidência de reações adversas ao medicamento (RAM) relatadas com Tarceva isolado ou em combinação com quimioterapia está resumida nas tabelas a seguir e é baseada nos dados de estudos clínicos.
As RAMs foram relatadas em pelo menos 10% dos pacientes (no grupo de Tarceva) e ocorreram mais frequentemente (? 3%) em pacientes tratados com Tarceva em relação ao braço comparador.
Informações adicionais de especial interesse das reações adversas
As seguintes reações adversas foram observadas em pacientes que receberam Tarceva 150 mg como monoterapia ou 100 mg ou 150 mg em combinação com gencitabina.
Composição
Cada comprimido revestido de 25 mg contém:
Erlotinibe (equivalente a 27,32 mg de cloridrato de erlotinibe) - 25 mg.
Cada comprimido revestido de 100 mg contém:
Erlotinibe (equivalente a 109,29 mg de cloridrato de erlotinibe) - 100 mg.
Cada comprimido revestido de 150 mg contém:
Erlotinibe (equivalente a 163,93 mg de cloridrato de erlotinibe) - 150 mg.
Excipientes: Lactose monoidratada, celulose microcristalina, amidoglicolato de sódio, laurilsulfato de sódio, estearato de magnésio.
Revestimento: hipromelose, hiprolose, dióxido de titânio e macrogol.
Superdosagem
Doses orais únicas de Tarceva de até 1.000 mg, em indivíduos saudáveis, e de até 1.600 mg, recebidas como dose única uma vez por semana, em pacientes com câncer, foram toleradas.
Doses repetidas duas vezes por dia, de 200 mg, em indivíduos saudáveis, foram mal toleradas após apenas alguns dias de administração.
Com base nos dados desses estudos, reações adversas graves, como diarreia, erupção cutânea e possivelmente elevação de transaminases hepáticas, podem ocorrer com a dose acima da recomendada.
Se você suspeitar de superdosagem, suspenda o uso de Tarceva e procure imediatamente o seu médico para que ele possa realizar o tratamento adequado.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou a bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de
mais orientações.
Interação Medicamentosa
Cloridrato de erlotinibe tem potencial para interações entre medicamentos clinicamente significativas.
Erlotinibe é metabolizado no fígado, por meio dos citocromos hepáticos humanos, principalmente o CYP3A4 e, em menor extensão, o CYP1A2 e a isoforma pulmonar CYP1A1. Potenciais interações podem ocorrer com fármacos que são metabolizados por essas enzimas ou são inibidores ou indutores delas.
Inibidores potentes de atividade do CYP3A4 reduzem o metabolismo de erlotinibe e aumentam as concentrações plasmáticas de erlotinibe. A inibição do metabolismo de CYP3A4 pelo cetoconazol (200 mg, VO, 2 vezes por dia, durante cinco dias) resultou em exposição aumentada do erlotinibe (86% em exposição mediana a erlotinibe [ASC]) e um aumento de 69% em Cmax quando comparado com erlotinibe apenas.
Quando cloridrato de erlotinibe foi coadministrado com ciprofloxacina, um inibidor de CYP3A4 e CYP1A2, a exposição de erlotinibe (ASC) e a concentração máxima (Cmáx) aumentaram em, aproximadamente, 39% e 17%, respectivamente. Portanto, deve-se ter cuidado ao administrar cloridrato de erlotinibe com inibidores potentes de CYP3A4 ou CYP3A4/CYP1A2 combinados. Nestas situações a dose de cloridrato de erlotinibe deve ser reduzida se for observada toxicidade.
Indutores potentes de atividade de CYP3A4 aumentam o metabolismo de erlotinibe e reduzem significativamente as concentrações plasmáticas de erlotinibe. A indução do metabolismo do CYP3A4 pela rifampicina (600 mg, VO, uma vez por dia, durante sete dias) resultou em redução de 69% na ASC mediana de erlotinibe, após uma dose de 150 mg de cloridrato de erlotinibe, em comparação com o uso de cloridrato de erlotinibe isolado.
O pré-tratamento e coadmnistração de rifampicina com uma dose única de 450 mg de cloridrato de erlotinibe resultou em exposição média de cloridrato de erlotinibe (ASC) de 57,5% daquela após uma dose única de 150 mg de cloridrato de erlotinibe na ausência do tratamento com rifampicina. Tratamentos alternativos com ausência de medicamentos com atividade indutora potente de CYP3A4 devem ser considerados, quando possível. Para pacientes que requerem tratamento concomitante de cloridrato de erlotinibe com um indutor potente da CYP3A4, tais como a rifampicina, deve-se considerar aumento para 300 mg na dose, com monitoramento rigoroso da segurança e, se bem tolerado por mais de duas semanas, pode se considerar aumento para 450 mg na dose, também com monitoramento rigoroso da segurança. Doses maiores não foram estudadas nesse cenário.
O pré-tratamento ou coadministração de cloridrato de erlotinibe não alterou a depuração dos substratos prototípicos de CYP3A4 midazolam e eritromicina. Portanto, são improváveis interações significativas na depuração de outros substratos do CYP3A4. A biodisponibilidade oral de midazolam pareceu diminuir em aproximadamente até 24% dos casos, entretanto, não se atribuiu aos efeitos na atividade do CYP3A4.
A solubilidade de erlotinibe é dependente do pH. A solubilidade de erlotinibe diminui com o aumento do pH. Medicamentos que alteram o pH do trato gastrintestinal superior podem alterar a solubilidade de erlotinibe e, por sua vez, sua biodisponibilidade. A coadministração de cloridrato de erlotinibe com omeprazol, um inibidor da bomba de próton, diminuiu a exposição de erlotinibe (ASC) e Cmáx em 46% e 61%, respectivamente. Não houve alterações no Tmáx ou meia-vida. A administração concomitante de cloridrato de erlotinibe com 300 mg de ranitidina, um antagonista do receptor H2, diminuiu a exposição de erlotinibe (ASC) e Cmáx em 33% e 54%, respectivamente. Portanto, a coadministração de medicamentos que reduzem a produção de ácido gástrico com cloridrato de erlotinibe deve ser evitada, quando possível. Aumento na dose de cloridrato de erlotinibe, quando coadministrado com tais agentes, não parece compensar essa perda de exposição. No entanto, quando cloridrato de erlotinibe foi administrado de forma a não coincidir, duas horas antes ou dez horas após, com a ranitidina, 150 mg, duas vezes ao dia, a exposição a erlotinibe (ASC e Cmáx) diminuíram apenas 15% e 17%, respectivamente. Caso os pacientes necessitem ser tratados com esses medicamentos, um antagonista do receptor H2, como a ranitidina, deve então ser considerado e usado de maneira a não coincidir os horários das doses. Cloridrato de erlotinibe deve ser ingerido duas horas antes ou dez horas após a ingestão de antagonista de receptores H2.
Interações com outros anticoagulantes cumarínicos, incluindo varfarina, que levaram a aumento da Razão Normatizada Internacional (INR) e eventos hemorrágicos, fatais em alguns casos, foram relatados em pacientes que receberam cloridrato de erlotinibe. Pacientes em uso de anticoagulantes derivados de cumarina devem ser monitorados regularmente em relação a alterações no tempo de protrombina ou na INR.
A combinação de cloridrato de erlotinibe com uma estatina pode aumentar o potencial de miopatia induzida por estatina, incluindo rabdomiólise, observada raramente.
Os fumantes devem ser aconselhados a parar de fumar, pois o cigarro, um indutor do CYP1A1 e CYP1A2, mostrou reduzir a exposição de erlotinibe em 50% – 60%.
Em um estudo de fase Ib, não houve efeito significante de gencitabina na farmacocinética de erlotinibe nem efeito significativo de erlotinibe na farmacocinética de gencitabina.
Ação da Substância
Resultados de Eficácia
Câncer de pulmão de não pequenas células (CPNPC)
Dados adicionais publicados
Em uma análise prospectiva dos pacientes com CPNPC avançado, que apresentavam tumores com mutações ativadoras no domínio TK do EGFR, a SLP mediana para os 113 pacientes tratados com Cloridrato De Erlotinibe (substância ativa) na primeira linha foi 14 meses (IC 95% 9,7 a 18,3 meses), e a mediana da sobrevida global foi 28,0 meses (IC 95%, 22,7 a 33 meses). A análise conjunta dos dados publicados a partir de pacientes com CPNPC demonstrou que pacientes com tumores com mutações ativadoras de EGFR tratados predominantemente com Cloridrato De Erlotinibe (substância ativa) na primeira linha (n=70; 12, 5 meses; IC 95% 10,6 a 16,0) apresentaram uma mediana maior de SLP, comparada com aqueles tratados com quimioterapia (n=359; 6,0 meses; IC 95% 5,4 a 6,7).
Terapia de manutenção de primeira linha
A eficácia e a segurança de Cloridrato De Erlotinibe (substância ativa) como terapia de manutenção de primeira linha em CPNPC foram demonstradas em estudo randomizado, duplo-cego e placebo controlado (BO18192). Esse estudo foi conduzido com 889 pacientes com CPNPC localmente avançado ou metastático que não progrediram durante 4 ciclos de quimioterapia baseada em esquemas duplos com platina. Os pacientes foram randomizados na proporção 1:1 com Cloridrato De Erlotinibe (substância ativa), 150 mg, ou placebo, por via oral, uma vez ao dia. O objetivo primário do estudo foi avaliar a sobrevida livre de progressão (SLP) em todos os pacientes e naqueles com tumor EGFR (receptor do fator de crescimento epidérmico) IHC (imunoistoquímica) positivo. As características demográficas e as características da doença foram bem equilibradas entre os dois braços de tratamento.
No estudo BO18192 (SATURN), a população geral demonstrou benefício para o desfecho primário de SLP (RR = 0,71 p<0,0001) e para o desfecho secundário de SG (RR = 0,81 p=0,0088). No entanto, o maior benefício foi observado em uma análise exploratória pré definida em pacientes com mutações ativadoras de EGFR (n=49), demonstrando um benefício substancial na SLP (RR = 0,10, IC 95%, 0,04 a 0,25; p<0,0001) e uma razão de risco de SG de 0,83 (IC 95%, 0,34 a 2,02). No subgrupo de mutações positivas de EGFR, 67% dos pacientes com placebo receberam segunda linha de tratamento ou linhas adicionais com inibidores de EGFR. Nos pacientes com tumores do tipo EGFR selvagem (n = 388), a razão de risco de SLP foi 0,78 (IC 95%, 0,63 a 0,96; p = 0,0185) e razão de risco de SG foi de 0,77 (IC 95%, 0,61 a 0,97; p = 0,0243).
O estudo BO25460 (IUNO) foi conduzido em 643 pacientes com câncer de pulmão de não pequenas células avançado cujos tumores não possuíam mutação ativadora de EGFR (deleção do exon 19 ou mutação no exon 21 L858R) e que não apresentaram progressão da doença após 4 ciclos de quimioterapia baseada em platina.
O objetivo do estudo foi comparar a SG da terapia de manutenção de primeira linha com erlotinibe versus erlotinibe administrado no momento da progressão da doença. O estudo não atingiu seu desfecho primário.
A SG de Cloridrato De Erlotinibe (substância ativa) na manutenção de primeira linha não foi superior à Cloridrato De Erlotinibe (substância ativa) no tratamento de segunda linha em pacientes cujo tumor não possuía mutação ativadora de EGFR (RR = 1,02, IC 95%, 0,85 a 1,22, p = 0,82). O desfecho secundário de SLP não apresentou diferença entre Cloridrato De Erlotinibe (substância ativa) e placebo no tratamento de manutenção (RR = 0,94, IC 95%, 0,80 a 1,11; p = 0,48).
Com base nos dados do estudo BO25460 (IUNO), Cloridrato De Erlotinibe (substância ativa) não é recomendado para o tratamento de manutenção de primeira linha em pacientes sem mutação ativadora de EGFR.
Terapia de segunda/ terceira linha
A eficácia e a segurança de Cloridrato De Erlotinibe (substância ativa) como terapia de segunda/ terceira linha foram demonstradas em estudo randomizado, duplo-cego, controlado com placebo (BR.21). Esse estudo foi conduzido em 17 países, incluindo 731 pacientes com CPNPC localmente avançado ou metastático, após falha de pelo menos um esquema quimioterápico. Os pacientes foram randomizados na proporção de 2:1 para receber Cloridrato De Erlotinibe (substância ativa) 150 mg ou placebo via oral diariamente. Os objetivos do estudo incluíram avaliar a sobrevida global, o tempo até deterioração de sintomas relacionados ao câncer de pulmão (tosse, dispneia e dor), taxa de resposta, duração da resposta, sobrevida livre de progressão (SLP) e segurança. O objetivo primário foi a sobrevida global.
Devido à randomização 2:1, 488 pacientes foram randomizados para Cloridrato De Erlotinibe (substância ativa) e 243 pacientes para placebo.
Os pacientes não foram selecionados por expressão imunohistoquímica HER1/EGFR, sexo, raça, história de tabagismo ou classificação histológica.
As características demográficas foram bem equilibradas entre os dois braços de tratamento.
Aproximadamente dois terços dos pacientes eram homens, um terço apresentava estado de desempenho ECOG basal de 2, e 9% dos pacientes apresentavam um ECOG basal de 3. Noventa e três por cento e 92% de todos os pacientes nos grupos Cloridrato De Erlotinibe (substância ativa) e placebo, respectivamente, tinham recebido um esquema prévio com platina, e 36% e 37% de todos os pacientes, respectivamente, tinham recebido uma terapia prévia com taxano. Cinquenta por cento dos pacientes tinham recebido apenas um esquema prévio de quimioterapia.
A sobrevida foi avaliada na população ITT. A mediana da sobrevida global aumentou em 42,5% e foi de 6,7 meses no grupo Cloridrato De Erlotinibe (substância ativa) (IC 95%, 5,5 a 7,8 meses), em comparação com 4,7 meses no grupo placebo (IC 95%, 4,1 a 6,3 meses), resultando em uma diferença de dois meses entre os grupos. A análise de sobrevida primária foi ajustada para fatores de estratificação, como descrito no momento da randomização (ECOG PS, melhor resposta à terapia prévia, número de esquemas prévios e exposição prévia à platina) e expressão imunohistoquímica HER1/EGFR. Nessa análise primária, a razão de risco ajustada para óbito no grupo Cloridrato De Erlotinibe (substância ativa) em relação ao grupo placebo foi de 0,73 (IC 95%, 0,60 a 0,87; p = 0,001). A porcentagem de pacientes vivos em 12 meses foi de 31,2% e 21,5%, respectivamente.
O benefício de sobrevida com o tratamento com Cloridrato De Erlotinibe (substância ativa) foi observado na maioria dos subgrupos. Uma série de subgrupos de pacientes formada pela estratificação das diferentes características do estado basal dos pacientes, tais como expressão imunohistoquímica HER1/EGFR, exposição prévia a taxanos, história de tabagismo, sexo, idade, histologia, perda de peso prévia, tempo entre o diagnóstico inicial e a randomização e localização geográfica, foi examinada por análises univariadas exploratórias para avaliar a consistência do resultado de sobrevida global. Praticamente todas as razões de risco (RR) no grupo Cloridrato De Erlotinibe (substância ativa), em relação ao grupo placebo, foram menores que 1,0, sugerindo que o benefício de Cloridrato De Erlotinibe (substância ativa) foi consistente entre os subgrupos. O benefício de sobrevida de Cloridrato De Erlotinibe (substância ativa) foi comparável em pacientes com performance status (PS) ECOG basal de 2 – 3 (RR = 0,77) ou um PS de 0 – 1 (RR = 0,73) e pacientes que receberam um regime de quimioterapia (RR = 0,76) ou dois ou mais regimes (RR=0,76).
O benefício de sobrevida de Cloridrato De Erlotinibe (substância ativa) também foi observado em pacientes que não atingiram uma resposta objetiva do tumor (critério RECIST). Isso foi evidenciado por uma razão de risco para óbito de 0,83 entre pacientes cuja melhor resposta foi doença estável e 0,85 entre pacientes cuja melhor resposta foi doença progressiva.
Em uma análise detalhada de alguns desses subgrupos, podemos explicitar as diferenças em meses de sobrevida entre os pacientes que receberam Cloridrato De Erlotinibe (substância ativa) ou placebo. No grupo de pacientes com ECOG 0 – 1, essa diferença foi de 1,45 mês (RR = 0,73) e com ECOG 2 – 3 de 0,39 mês (RR = 0,77); no grupo de pacientes que havia recebido um tratamento quimioterápico prévio, a diferença foi de 0,83 mês (RR = 0,76) e de 2,18 meses (RR=0,76) no grupo que havia recebido dois ou mais tratamentos prévios.
Entre os pacientes que nunca fumaram e que foram tratados com Cloridrato De Erlotinibe (substância ativa), o ganho de sobrevida foi mais que seis meses: a sobrevida mediana no grupo de pacientes não fumantes tratados com Cloridrato De Erlotinibe (substância ativa) foi de 12,25 meses e no grupo de pacientes não fumantes que receberam placebo foi de 5,62 meses.
Não se pode afirmar que houve benefício entre pacientes fumantes ou ex-fumantes: a sobrevida mediana obtida no grupo de pacientes fumantes ou ex-fumantes que foram tratados com Cloridrato De Erlotinibe (substância ativa) foi de 5,52 meses e no grupo de pacientes fumantes ou ex-fumantes que receberam placebo foi de 4,63 meses, resultando em diferença de 0,89 mês.
Câncer de pâncreas (Cloridrato De Erlotinibe (substância ativa) administrado simultaneamente com gencitabina)
A eficácia e a segurança de Cloridrato De Erlotinibe (substância ativa) em combinação com gencitabina como tratamento de primeira linha foram avaliadas em um estudo randomizado, duplo-cego, placebo controlado, em 569 pacientes com câncer de pâncreas localmente avançado, irressecável ou metastático. Os pacientes foram randomizados na proporção de 1:1 para receber Cloridrato De Erlotinibe (substância ativa) (100 ou 150 mg) ou placebo, uma vez ao dia, em esquema contínuo, mais gencitabina IV (1.000 mg/m2, Ciclo 1 – Dias 1, 8, 15, 22, 29, 36 e 43 de um ciclo de oito semanas, Ciclo 2 e ciclos subsequentes – Dias 1, 8 e 15 de um ciclo de 4 semanas - dose aprovada e esquema para câncer de pâncreas, vide bula de gencitabina). Cloridrato De Erlotinibe (substância ativa) ou placebo foi ingerido uma vez ao dia até a progressão da doença ou toxicidade inaceitável. O objetivo do estudo incluiu sobrevida global, taxa de resposta e sobrevida livre de progressão (SLP). A duração da resposta também foi avaliada. O objetivo primário foi sobrevida. Um total de 285 pacientes foi randomizado para receber gencitabina mais Cloridrato De Erlotinibe (substância ativa) (261 pacientes do grupo de 100 mg e 24 pacientes do grupo de 150 mg) e 284 pacientes foram randomizados para receber gencitabina mais placebo (260 pacientes do grupo de 100 mg e 24 pacientes no grupo de 150 mg). O pequeno número de pacientes na dose de 150 mg não permitiu conclusões.
Características basais demográficas e da doença dos pacientes foram similares entre os dois grupos de tratamento, Cloridrato De Erlotinibe (substância ativa) 100 mg mais gencitabina ou placebo mais gencitabina, exceto para uma proporção levemente aumentada de mulheres no braço Cloridrato De Erlotinibe (substância ativa) (51%) comparado com o braço placebo (44%). O tempo mediano do diagnóstico inicial para a randomização foi aproximadamente um mês. Aproximadamente metade dos pacientes teve performance status (PS) ECOG basal de 1, e 17% tiveram ECOG PS basal de 2. A maioria dos pacientes apresentou-se com doença metastática na entrada do estudo como manifestação inicial do câncer de pâncreas (77% no braço Cloridrato De Erlotinibe (substância ativa), 76% no braço placebo).
A sobrevida foi avaliada na população com intenção de tratamento com base nos dados de acompanhamento de sobrevida, incluindo 551 óbitos. Os resultados são apresentados para o grupo de dose de 100 mg (504 óbitos). A razão de risco ajustada para óbito no grupo Cloridrato De Erlotinibe (substância ativa) relativo ao grupo placebo foi 0,82 (IC 95% 0,69 a 0,98; p = 0,028). A porcentagem de pacientes vivos aos 12 meses foi de 23,8% no grupo Cloridrato De Erlotinibe (substância ativa) comparado aos 19,4% no grupo placebo. O tratamento com Cloridrato De Erlotinibe (substância ativa) esteve associado a discreto ganho de sobrevida global, estatisticamente significante. A sobrevida global mediana foi de 6,4 meses no grupo Cloridrato De Erlotinibe (substância ativa) comparado com 6 meses no grupo placebo.
A Tabela 2 resume os resultados do estudo.
Tabela 2: Resultados de eficácia do estudo PA.3
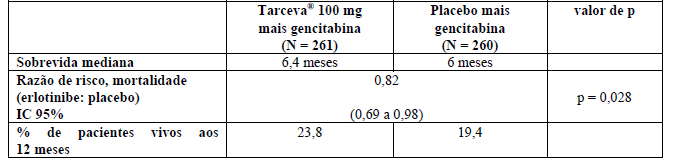
A SLP mediana foi de 3,81 meses (16,5 semanas) no grupo Cloridrato De Erlotinibe (substância ativa) (IC 95%, 3,58 a 4,93 meses), comparado com 3,55 meses (15,2 semanas) no grupo placebo (IC 95% 3,29 a 3,75 meses; p = 0,006).
A duração mediana de resposta foi de 23,9 semanas, intervalo de 3,71 a 56+ semanas. A taxa de resposta objetiva (resposta completa e resposta parcial) foi de 8,6% no grupo Cloridrato De Erlotinibe (substância ativa) e 7,9% no grupo placebo. A proporção de pacientes que apresentaram resposta completa, resposta parcial ou doença estável foi de 59% e 49,4%, respectivamente, para os grupos Cloridrato De Erlotinibe (substância ativa) e placebo (p = 0,036).
Caractéristicas Farmacológicas
Farmacodinâmica
Erlotinibe potencialmente inibe a fosforilação intracelular do receptor HER1/EGFR. O receptor HER1/EGFR é expresso na superfície celular de células normais e de células cancerosas. Nos modelos não clínicos, a inibição da fosforilação do EGFR resulta em inibição da proliferação celular e/ou morte celular.
Farmacocinética
Após a dose oral de Cloridrato De Erlotinibe (substância ativa) 150 mg, o tempo mediano para alcançar o estado de equilíbrio é de quatro horas, com a concentração plasmática mediana máxima em torno de 1,995 ng/mL. Antes da próxima dose em 24 horas, a concentração plasmática mediana mínima é de 1,238 ng/mL. A mediana da ASC observada durante os intervalos de dose no estado de equilíbrio é 41,300 g*h/mL.
Farmacocinética em populações especiais
Não houve nenhum estudo específico em pacientes de faixa etária pediátrica ou em idosos.
Segurança pré-clínica
Cuidados de Armazenamento
Você deve conservar Tarceva em temperatura ambiente (entre 15 e 30 ºC).
Número de lote e datas de fabricação e validade: Vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Descarte de medicamentos não utilizados e / ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado.
Utilize o sistema de coleta local estabelecido, se disponível.
Caracterísitca física:
Os comprimidos revestidos de Tarceva são biconvexos, de cor branca a amarelada.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS – 1.0100.0651
Farm. Resp.:
Tatiana Tsiomis Díaz – CRF-RJ nº 6942
Fabricado na Suíça por:
F.Hoffmann-La Roche Ltd, Basileia
ou Fabricado para F.Hoffmann-La Roche Ltd, Basileia, Suíça
por Roche S.p.A., Segrate, Itália
Embalado por:
F. Hoffmann-La Roche Ltd, Kaiseraugst, Suíça
Registrado, importado e distribuído no Brasil por:
Produtos Roche Químicos e Farmacêuticos S.A.
Est. dos Bandeirantes, 2.020 – CEP 22775-109 – Rio de Janeiro – RJ
CNPJ 33.009.945/0023-39
Serviço Gratuito de Informações – 0800 7720 289
www.roche.com.br
Venda sob prescrição médica.
informações complementares
| Fabricante |
| ROCHE |
| Princípio ativo |
| Erlotinibe |
| Categoria do medicamento |
| Medicamentos Especiais |
TARCEVA 100MG 30 COMPRIMIDOS É UM MEDICAMENTO, NÃO USE SEM PRESCRIÇÃO MÉDICA E ORIENTAÇÃO DO FARMACÊUTICO. AO PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO.