- Medicamentos
- Medicamentos Especiais
- Zelboraf - 240Mg 56 Comprimidos
Comparamos o preço de zelboraf - 240mg 56 comprimidos, veja o menor preço
Zelboraf - 240Mg 56 Comprimidos

Menor Preço
R$ 10.590,00
- CATEGORIA: Medicamentos Especiais
- PRINCÍPIO ATIVO: Vemurafenibe
- FABRICANTE: ROCHE
PARA QUE SERVE?
Para que serve Zelboraf é indicado para o tratamento de melanoma (doença maligna das células da pele que produzem o pigmento chamado melanina) que não possa ser retirado ou que apresente metástases. Está indicado para casos de melanoma que apresentem um tipo de mutação chamada de BRAF V600E, quando detectado por um teste aprovado pela ANVISA.
R
Referência
16
ofertasMelhores preços a partir de R$ 10.590,00 até R$ 14.400,70
Menor preço
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Onco Express Medicamentos Especiais e Oncológicos
economize
18.59%
R$ 11.724,28
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Preço Válido para compra em Boleto
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Para que serve
Zelboraf é indicado para o tratamento de melanoma (doença maligna das células da pele que produzem o pigmento chamado melanina) que não possa ser retirado ou que apresente metástases. Está indicado para casos de melanoma que apresentem um tipo de mutação chamada de BRAF V600E, quando detectado por um teste aprovado pela ANVISA.
Como Zelboraf funciona?
Zelboraf é uma molécula que inibe a ação de algumas enzimas no tumor, diminuindo a multiplicação e a sobrevida de suas células.
O tempo mediano observado para resposta ao tratamento com Zelboraf foi de 1,45 mês (variação de 1,0 a 5,5).
Contraindicação
Você não deverá usar Zelboraf se tiver alergia ao princípio ativo (vemurafenibe) ou às substâncias usadas na fabricação do comprimido revestido.
Como usar
A dose recomendada de Zelboraf é de 960 mg (quatro comprimidos de 240 mg), duas vezes por dia, totalizando oito comprimidos ao dia.
A primeira dose deve ser tomada pela manhã, e a segunda, à noite, com um intervalo de, aproximadamente, 12 horas entre as duas doses. As doses podem ser tomadas com ou sem alimentos.
Os comprimidos devem ser engolidos com um copo de água. Os comprimidos não podem ser esmagados nem mastigados.
O tratamento deverá continuar até que apareça progressão da doença ou até que apareça efeito colateral intolerável.
Se ocorrerem efeitos colaterais da medicação que provoquem sintomas ou o aumento do intervalo QT no eletrocardiograma, pode ser necessário reduzir a dose ou descontinuar Zelboraf. Não são recomendáveis reduções para menos de 480 mg duas vezes por dia. Em caso de câncer de pele tipo espinocelular, não são recomendadas reduções da dose de Zelboraf.
Caso haja vômitos após a administração de Zelboraf, você não deve tomar uma dose adicional do medicamento e o tratamento deve continuar como de costume.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que eu devo fazer quando eu me esquecer de usar Zelboraf?
Se você esquecer (ou não puder por qualquer motivo) de tomar uma das doses, ela poderá ser tomada até quatro horas antes da dose seguinte. Você não poderá dobrar a dose seguinte para compensar uma dose perdida.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Você só deve tomar Zelboraf se apresentar tumor com mutação BRAF V600E confirmada.
Alguns pacientes que estavam tomando Zelboraf desenvolveram um câncer de pele chamado espinocelular cutâneo. Esse câncer ocorreu, geralmente, no início do tratamento e com maior frequência em pessoas com 65 anos ou mais, com histórico de câncer de pele anteriormente e com exposição crônica ao sol. Em geral, é possível tratar esse tipo de câncer retirando a lesão, e o tratamento com Zelboraf pode ser mantido. No entanto, é recomendável que você faça um exame dermatológico antes de começar a tomar Zelboraf e continue fazendo exames periódicos durante o tratamento e até seis meses depois de terminá-lo. Se você notar qualquer lesão cutânea, avise ao seu médico.
Têm sido reportados casos de câncer espinocelular que não fossem cutâneos em pacientes recebendo Zelboraf.
Você deverá fazer exames de cabeça e pescoço antes de iniciar o tratamento e a cada três meses durante o tratamento. Além disso, deverá fazer uma tomografia de tórax antes do início do tratamento e a cada seis meses a partir de então. Exames pélvicos (para mulheres) e exames anais são recomendados antes e no final do tratamento ou conforme orientação médica.
Novos melanomas primários têm sido reportados em estudos clínicos. Os casos foram tratados e os pacientes continuaram utilizando Zelboraf sem necessidade de alterar a dose. Você deverá ser monitorado por seu médico, conforme descrito acima para câncer espinocelular cutâneo.
Zelboraf pode causar progressão de cânceres associados com mutações do gene RAS (exacerbação de caso de leucemia crônica foi reportado). Zelboraf deve ser usado com cautela em pacientes com histórico de câncer associado à mutação RAS ou que passe a apresentá-lo durante o tratamento.
Foram relatados casos de reações alérgicas graves, incluindo anafilaxia (reação grave, geralmente imediata, que inclui queda abrupta da pressão arterial e dificuldade respiratória), lesões de pele generalizadas e vermelhidão em todo o corpo com queda da pressão arterial. Se isso ocorrer no seu caso, você não poderá utilizar Zelboraf novamente.
Reações dermatológicas graves têm sido reportadas por pacientes em tratamento com Zelboraf, incluindo casos raros de síndrome de Stevens-Johnson (inclui lesões cutâneas generalizadas, como bolhas, que podem atingir também as mucosas), necrólise epidérmica tóxica (camada superficial da pele se solta em lâminas) e reação ao medicamento com eosinofilia e sintomas sistêmicos (síndrome Dress) (síndrome de reação ao medicamento associada à elevação de células do sangue e sintomas pelo corpo). Se isso ocorrer no seu caso, você não poderá utilizar Zelboraf novamente.
Casos de dermatite (inflamação de pele) relacionada à radiação e sensibilização à radiação foram reportados em pacientes tratados com radiação antes, durante ou após o tratamento com Zelboraf. A maioria dos casos de sensibilização foi relacionada à pele, mas alguns casos envolveram órgãos internos e foram fatais. Zelboraf deve ser usado com cautela quando administrado concomitantemente ou após o tratamento com radiação.
Em um estudo com Zelboraf, foi constatado prolongamento do intervalo entre as ondas Q e T (comumente chamado de intervalo QT) do eletrocardiograma. Isso pode levar a arritmias do coração, que podem ser graves.
Por isso, se você estiver com alterações dos eletrólitos no sangue (por exemplo, sódio, potássio, cálcio e magnésio) que não possam ser corrigidas, tiver diagnóstico de QT longo ou estiver recebendo medicamentos que prolonguem o intervalo QT, não deverá utilizar Zelboraf.
Além disso, será necessário monitorar o seu eletrocardiograma e os eletrólitos em seu sangue durante o tratamento com Zelboraf e sempre que a dose deste medicamento for alterada.
Pode haver necessidade de diminuir a dose de Zelboraf ou mesmo de suspender o tratamento de acordo com as alterações encontradas.
Lesão no fígado, incluindo casos de lesão grave, foi relatada com o uso de Zelboraf. Podem ocorrer alterações dos exames laboratoriais referentes ao fígado durante o tratamento com Zelboraf. Por isso, é necessário medir as enzimas hepáticas (exame de sangue que dosa enzimas do fígado, para ver se está ocorrendo lesão das células do fígado) antes do início do tratamento e monitorá-las mensalmente durante o tratamento, conforme orientação médica.
Se houver alteração, poderá ser necessário reduzir a dose ou suspender o medicamento.
Os dados de segurança e eficácia dos estudos clínicos de Zelboraf foram coletados em pacientes utilizando o medicamento com ou sem alimentos.
Pode haver reação de fotossensibilidade (quando aparecem reações na pele ao se expor à luz) de intensidade leve a grave. Você deverá evitar exposição solar enquanto estiver recebendo Zelboraf e por, no mínimo, cinco dias após o término do tratamento, usando roupas que protejam do sol e filtro solar UVA e UVB, juntamente com protetor labial (Fator de Proteção Solar – FPS – maior que 30).
A contratura de Dupuytren (contratura fixa da mão em flexão caracterizada pelo espessamento da fáscia palmar – tecido fibroso no interior das palmas das mãos) e a fibromatose plantar (tumoração endurecida na planta dos pés, que pode ou não ser dolorosa) foram relatadas com Zelboraf. A maioria dos casos foram de graus leve a moderado, mas também foram notificados casos graves e incapacitantes de contratura de Dupuytren.
Caso você apresente algum desses eventos, informe seu médico, pois alterações em seu tratamento podem ser necessárias.
Reações graves nos olhos, incluindo uveíte (inflamação no olho), foram relatadas. Os pacientes devem ser monitorados rotineiramente em relação às reações nos olhos.
Não é recomendada a administração de ipilimumabe e Zelboraf ao mesmo tempo.
Podem ocorrer anormalidades laboratoriais relacionadas à creatinina (exame de sangue feito para avaliar o funcionamento dos rins). Por isso, creatinina sérica deve ser medida antes do início do tratamento e periodicamente monitorada durante o tratamento, conforme indicação médica.
Não foram feitos estudos ainda sobre a influência deste medicamento na capacidade de dirigir veículos ou operar máquinas.
Não foram feitos estudos em gestantes, no entanto, a transferência placentária de vemurafenibe para o feto foi reportada. Baseado em seu mecanismo de ação, o vemurafenibe pode causar dano fetal quando administrado a uma mulher grávida. Embora não tenham sido constatadas malformações em estudos feitos com animais de laboratório, não é possível garantir a segurança à mulher grávida.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Uso na gravidez apenas se os potenciais benefícios para a mãe superarem os possíveis riscos para o feto.
Não foram realizados estudos pré-clínicos de fertilidade com Zelboraf.
Mulheres com possibilidade de engravidar e homens devem usar medidas contraceptivas adequadas durante o tratamento com Zelboraf e durante, pelo menos, seis meses depois do término do tratamento.
Não se sabe se Zelboraf é seguro no trabalho de parto ou no parto.
Não foi ainda estabelecido se Zelboraf pode ser passado para o leite materno. Deve-se tomar uma decisão entre descontinuar a amamentação ou Zelboraf depois de considerar os benefícios do aleitamento materno para a criança e os benefícios da terapia para a mãe.
A segurança e a eficácia de Zelboraf em crianças e adolescentes com menos de 18 anos ainda não foram estabelecidas.
Estudos realizados em pessoas com 65 anos ou mais mostraram que os efeitos de Zelboraf são semelhantes aos que ocorrem nos adultos mais jovens e que não é necessário fazer ajustes na dose.
Os eventos adversos mais sérios que ocorreram com maior frequência em mulheres que em homens foram: reações de pele, dor nas articulações e fotossensibilidade.
Os dados relativos ao tratamento com Zelboraf, em pacientes com funcionamento dos rins ou fígado prejudicado, são limitados. O risco de aumento de exposição de Zelboraf em pacientes com funcionamento dos rins e fígado gravemente prejudicado não pode ser excluído.
Até o momento, não há informações de que Zelboraf possa causar doping. Em caso de dúvida, consulte o seu médico.
O uso de Zelboraf pode aumentar a quantidade de cafeína, varfarina (remédio usado para diminuir a coagulação do sangue), tizanidina (remédio usado como relaxante muscular) e dextrometorfano (remédio usado para aliviar a tosse) e diminuir a de midazolam (remédio para dormir), se você estiver usando esses medicamentos durante o tratamento. Zelboraf é um inibidor moderado de CYP1A2 e um indutor de CYP3A4 (enzimas essenciais no metabolismo de muitos medicamentos).
Zelboraf pode aumentar a concentração no sangue de medicamentos predominantemente metabolizados por CYP1A2 e diminuir a concentração no sangue de medicamentos metabolizados predominantemente por CYP3A4.
Zelboraf é um inibidor da glicoproteína P (transportador que promove ativamente a saída da substância ativa do interior da célula). Zelboraf pode aumentar a concentração no sangue de medicamentos que são substratos da glicoproteína P, como por exemplo, digoxina e clopidrogrel.Se estiver usando algum desses medicamentos informe seu médico.
A administração concomitante de Zelboraf com um forte inibidor de CYP3A4 (ex.: cetoconazol, itraconazol, claritromicina, atazanavir, nefazodona, saquinavir, telitromicina, ritonavir, indinavir, nelfinavir e voriconazol) ou indutor de CYP3A4 (ex.: fenitoína, carbamazepina, rifampicina, rifabutina, rifapentina e fenobarbital) pode alterar as concentrações de Zelboraf.
Pacientes em tratamento com Zelboraf apresentam maior potencial de toxicidade ao tratamento com radiação.
Deve-se ter cautela ao administrar Zelboraf juntamente com digoxina (medicamento utilizado para mau funcionamento do coração), pois Zelboraf pode alterar as concentrações de digoxina.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
As reações adversas mais frequentes de qualquer grau foram artralgia (dor nas articulações), fadiga (cansaço), erupção cutânea (reação na pele), fotossensibilidade (sensibilidade aumentada a luz), alopecia (queda de pelos ou cabelo), náuseas (enjoo), diarreia, cefaleias, prurido, vômitos, papiloma cutâneo e hiperqueratose (tipos de lesões na pele).
As reações adversas de grau 3 mais comuns foram carcinoma espinocelular cutâneo (tumor de pele), ceratoacantoma (tipo de lesão de pele), erupção cutânea, artralgia e aumento na gama-glutamiltransferase (GGT – uma enzima do fígado).
Reações adversas que ocorreram em pacientes com melanoma irressecável ou metástico
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento)
Distúrbios de pele e do tecido subcutâneo:
Erupção (lesões) de pele, reações de fotossensibilidade (sensibilidade à luz), alopecia (queda de pelos ou cabelos), coceira, hiperqueratose (formação de pele anormalmente grossa, como um calo), exantema maculopapular (formação de lesões elevadas na pele), queratose actínica (espessamento anormal da pele), pele seca, eritema (vermelhidão), síndrome de eritrodisestesia palmoplantar (sintomas de vermelhidão e
sensações anormais em palmas e plantas) e queratose pilar (aumento da espessura da pele próximo aos pelos, vulgarmente “bolinhas”).
Distúrbios musculoesqueléticos e do tecido conectivo:
Artralgia (dor nas articulações, “juntas”), mialgia (dor muscular), dor nos membros, dor musculoesquelética (dor nos músculos e ossos ou articulações), dor nas costas, artrite (inflamação de articulações).
Distúrbios gerais e condições de administração:
Fadiga (sensação de cansaço generalizado), edema (inchaço) nos membros, febre, astenia (sensação de fraqueza muscular generalizada).
Distúrbios gastrintestinais:
Náuseas (enjoo), diarreia, vômitos, constipação (intestino “preso”).
Distúrbios do sistema nervoso:
Cefaleia (dor de cabeça), disgeusia (alteração do paladar), neuropatia periférica (afecção dos nervos de mãos e pés) e vertigem (tontura).
Neoplasias benignas, malignas e não especificadas (incluindo cistos e pólipos) :
Papiloma cutâneo (tumor benigno da pele), carcinoma espinocelular de pele (câncer de pele), queratoacantoma e queratose seborreica (lesões de pele).
Distúrbios hepatobiliares:
Aumento da gama-glutamiltransferase (enzima do fígado).
Distúrbios do metabolismo e nutrição:
Redução do apetite e redução de peso.
Distúrbios respiratórios, torácicos e mediastinais:
Tosse.
Lesões, envenenamentos e complicações de procedimentos:
Queimadura solar.
Infecções e Infestações:
Foliculite (inflamação no local onde nascem os pelos).
Reação comum (ocorre entre 1% a 10% dos pacientes que utilizam este medicamento)
Distúrbios de pele e do tecido subcutâneo:
Exantema papular (tipo de lesão da pele), paniculite (inflamação com formação de nódulos sob a pele) e eritema nodoso (lesões avermelhadas como pequenos tumores dolorosos sob a pele).
Distúrbios do sistema nervoso:
Paralisia do sétimo nervo (paralisia de nervo da face).
Neoplasias benignas, malignas e não especificadas (incluindo cistos e pólipos):
Carcinoma basocelular (tipo de tumor de pele).
Distúrbios cardíacos:
Prolongamento do intervalo QT do eletrocardiograma.
Distúrbios oftalmológicos:
Uveíte (inflamação do olho) e iridociclite (inflamação da parte anterior do olho).
Distúrbios vasculares:
Vasculite (inflamação dos vasos).
Reação incomum (ocorre entre 0,1% a 1% dos pacientes que utilizam este medicamento)
Distúrbios de pele e do tecido subcutâneo:
Síndrome de Stevens-Johnson (lesões, como bolhas na pele e nas mucosas) e necrólise epidérmica tóxica(camada superficial da pele se solta em lâminas).
Distúrbios musculoesqueléticos e do tecido conectivo:
Contratura de Dupuytren (contratura fixa da mão em flexão caracterizada pelo espessamento da fáscia palmar – tecido fibroso no interior das palmas das mãos).
Distúrbios oftalmológicos:
Oclusão (entupimento) da veia da retina.
Mais informações sobre reações adversas selecionadas:
Carcinoma espinocelular cutâneo (CEC):
Nos estudos com pacientes com melanoma irressecável ou metastático, a incidência de CEC em pacientes tratados com Zelboraf foi de, aproximadamente, 20%. A maioria foi classificada como subtipos mais benignos e menos invasivos de CEC.
A maioria das lesões classificadas como “outras” (43%) era lesão benigna (por exemplo: verruga vulgar, queratose actínica, queratose benigna, cisto / cisto benigno).
Reações de hipersensibilidade:
Foi reportado um caso de reação de hipersensibilidade, com erupção cutânea, febre, tremores e queda da pressão arterial oito dias depois do início do tratamento com 960 mg de Zelboraf, duas vezes por dia, em um estudo clínico. Sintomas similares foram observados com a reintrodução do tratamento com uma dose única de 240 mg de Zelboraf. O paciente descontinuou definitivamente Zelboraf e se recuperou sem sequelas.
Prolongamento de QT:
A análise de dados eletrocardiográficos de um estudo mostrou que ocorre um aumento médio do intervalo entre a onda Q e a onda T no eletrocardiograma em relação ao valor normal do paciente. Esse prolongamento aumenta com a dose e com o tempo de tratamento até o 15º dia.
A partir do primeiro mês de tratamento, permanece estável. A previsão é de que o prolongamento pode ser maior para pacientes obesos (com IMC de 45 kg/m2) e ocorrer com maior frequência em mulheres.
Alterações laboratoriais
Ocorreram alterações laboratoriais hepáticas nos estudos realizados com Zelboraf em pacientes com melanoma irressecável ou metastático em gama-glutamiltransferase (GGT) (11,5%), aspartato aminotrasferase (AST) (0,9%), alanina aminotransferase (ALT) (2,8%), fosfatase alcalina (2,9%) e bilirrubinas (1,9%). Exceto para GGT e AST, nenhum paciente apresentou alterações grau 4. Também podem ocorrer alterações de creatinina (27,9%).
Experiência pós-comercialização
Foram relatados casos de exacerbação de leucemia crônica (LMMC)*, adenocarcinoma pancreático (tumor no pâncreas)*, síndrome de reação ao medicamento associada à elevação de células do sangue e sintomas pelo corpo (síndrome Dress), danos causados por radiação (inflamação e danos na pele, inflamação dos pulmões, do esôfago, do reto, do fígado, da bexiga e morte de tecidos), lesão aguda nos rins, contratura de Dupuytren (contratura fixa da mão em flexão caracterizada pelo espessamento da fáscia palmar ) e fibromatose plantar (tumoração endurecida na planta dos pés, que pode ou não ser dolorosa), todos com frequência desconhecida. Relatos de lesão no fígado, neutropenia (diminuição de células brancas no sangue) e pancreatite (inflamação do pâncreas) foram incomuns.
* Leucemia mielomonocítica crônica e adenocarcinoma pancreático preexistentes, ambos com mutação n-ras.
Alterações laboratoriais
No período pós-comercialização, foram reportadas alterações laboratoriais de fígado e elevação simultânea da concentração de bilirrubina. Houve também alterações laboratoriais de creatinina.
Atenção: Este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
Composição
Cada comprimido contém:
240 mg de vemurafenibe na forma de dispersão sólida em hipromelose acetato succinato.
Excipientes: Dióxido de silício, croscarmelose sódica, hiprolose e estearato de magnésio. Componentes do revestimento: álcool polivinílico, dióxido de titânio, macrogol, talco e óxido férrico vermelho.
Superdosagem
Não existe nenhum antídoto para Zelboraf. Se você apresentar efeitos colaterais, precisará receber tratamento para os sintomas. Se aparecer lesão de pele grave com muita coceira e cansaço, você precisa suspender Zelboraf imediatamente e entrar em contato com seu médico.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Efeitos de Vemurafenibe (substância ativa) em enzimas metabolizadoras de medicamentos
Resultados de um estudo in vivo de interação entre medicamentos em pacientes com melanoma metastático demonstraram que Vemurafenibe (substância ativa) é um inibidor moderado de CYP1A2 e indutor de CYP3A4.
Não é recomendado o uso concomitante de Vemurafenibe (substância ativa) com agentes metabolizados pelo CYP1A2 e CYP3A4 que possuam janelas terapêuticas estreitas. Se a administração concomitante não puder ser evitada, é necessário ter cautela, uma vez que Vemurafenibe (substância ativa) pode aumentar a exposição plasmática de medicamentos contendo substratos CYP1A2 e diminuir a exposição plasmática de medicamentos contendo substratos CYP3A4. Se clinicamente indicada, redução de dose do medicamento concomitante contendo substrato CYP1A2 deve ser considerada. A coadministração de Vemurafenibe (substância ativa) aumentou a AUC da cafeína (substrato CYP1A2) 2,6 vezes, enquanto decresceu a AUC de midazolam (substrato CYP3A4) em 39% nos estudos clínicos. Em outro estudo clínico, Vemurafenibe (substância ativa) aumentou AUCúltima e AUCinf de uma dose única de 2 mg de tizanidina (um substrato CYP1A2) em, aproximadamente, 4,2 e 4,7 vezes, respectivamente.
A AUC de dextrometorfano (substrato CYP2D6) e de seu metabólito dextrorfano aumentaram aproximadamente 47%, indicando um efeito sobre a cinética de dextrometorfano que talvez não possa ser mediada pela inibição de CYP2D6.
A administração concomitante de Vemurafenibe (substância ativa) resultou em um aumento de 18% na AUC de S-varfarina (substrato CYP2C9). É necessário ter cautela e considerar monitoramento RNI (Razão Normalizada Internacional) adicional quando Vemurafenibe (substância ativa) é administrado concomitantemente com varfarina.
Vemurafenibe (substância ativa) inibe moderadamente o CYP2C8 in vitro. A relevância in vivo desse achado é desconhecida, mas o risco de um efeito clinicamente relevante na administração concomitantemente com substratos do CYP2C8 não pode ser excluído. A administração concomitante de substratos do CYP2C8 com uma janela terapêutica estreita deve ser feita com cautela, pois Vemurafenibe (substância ativa) pode aumentar as concentrações desses substratos.
Medicamentos que inibem ou induzem CYP3A4
Vemurafenibe (substância ativa) é um substrato de CYP3A4 e, portanto, a administração concomitante de um forte inibidor ou indutor de CYP3A4 pode alterar as concentrações de Vemurafenibe (substância ativa). A coadministração de rifampicina, um forte indutor de CYP3A4, diminuiu significativamente a exposição plasmática de vemurafenibe (AUC) em, aproximadamente, 40% após uma dose única de 960 mg de Vemurafenibe (substância ativa). Fortes inibidores de CYP3A4 (ex.: cetoconazol, itraconazol, claritromicina, atazanavir, nefazodona, saquinavir, telitromicina, ritonavir, indinavir, nelfinavir e voriconazol) e indutores (ex.: fenitoína, carbamazepina, rifampicina, rifabutina, rifapentina e fenobarbital) devem ser utilizados com cautela quando administrados concomitantemente ao Vemurafenibe (substância ativa).
Tratamento com radiação
Potencialização de toxicidade relacionada ao tratamento com radiação foi reportada em pacientes recebendo Vemurafenibe (substância ativa). Na maioria dos casos, pacientes recebendo regimes radioterápicos maiores ou iguais a 2 Gy/dia (regimes hipofracionados).
Interação de Vemurafenibe (substância ativa) com sistema de transporte de medicamentos
Estudos in vitro demonstraram que Vemurafenibe (substância ativa) é tanto um substrato quanto um inibidor do transportador de efluxo, glicoproteína P (P-gp) e da proteína resistente ao câncer de mama (BCRP).
O estudo clínico de interação medicamentosa GO28394 utilizando medicamento contendo substrato P-gp (digoxina) demonstrou que doses orais múltiplas de Vemurafenibe (substância ativa) (960 mg, duas vezes ao dia) aumentam a exposição de uma dose oral única de digoxina, com um aumento de, aproximadamente, 1,8 e 1,5 vezes da AUCúltima e Cmáx de digoxina, respectivamente. Deve-se ter cautela quando administrar Vemurafenibe (substância ativa) concomitantemente com substratos P-gp. Deve ser considerada redução de dose do medicamento concomitante contendo substrato P-gp, se clinicamente indicado.
Os efeitos de Vemurafenibe (substância ativa) sobre medicamentos que sejam substratos de BCRP e os efeitos de indutores e inibidores de BCRP sobre exposição a Vemurafenibe (substância ativa) são desconhecidos.
Estudos in vitro também demonstraram que Vemurafenibe (substância ativa) é um inibidor da bomba de exportação de sais biliares. A relevância desse achado in vivo é desconhecida.
Interação Alimentícia
Alimentos (refeição rica em gordura) aumentam a biodisponibilidade relativa de uma dose única de 960 mg de Vemurafenibe (substância ativa).
As razões geométricas médias entre os estados alimentado e em jejum para Cmáx e AUC foram 2,5 e 4,6 à 5,1 vezes, respectivamente.
O Tmáx mediano aumentou de 4 para 7,5 horas quando uma dose única de Vemurafenibe (substância ativa) foi administrada com alimentos. Nos estudos pivotais, os dados de segurança e eficácia foram coletados em pacientes utilizando Vemurafenibe (substância ativa) com ou sem alimentos.
Ação da Substância
Resultados de Eficácia
A eficácia de Vemurafenibe (substância ativa) foi avaliada em 675 pacientes de um estudo clínico Fase III1 e 132 pacientes de um estudo clínico Fase II.2 Antes da inclusão no estudo, amostras de tumores de todos os pacientes foram testadas para verificar a presença de uma mutação BRAF V600 pelo teste de mutação cobas 4800 BRAF V600.
Pacientes sem Tratamento Prévio
Um estudo Fase III, aberto, multicêntrico, internacional, randomizado apoia o uso de Vemurafenibe (substância ativa) em pacientes sem tratamento prévio que apresentem melanoma irressecável ou metastático positivo para mutação BRAF V600. Os pacientes foram randomizados para tratamento com Vemurafenibe (substância ativa) (960 mg, duas vezes ao dia) ou dacarbazina (1.000 mg/m2 a cada três semanas).
No total, 675 pacientes foram randomizados para Vemurafenibe (substância ativa) (n = 337) ou dacarbazina (n = 338). A randomização foi estratificada de acordo com o estágio da doença, DHL, classificação ECOG e região geográfica.
As características iniciais foram bem equilibradas entre os grupos de tratamento. Para pacientes randomizados para Vemurafenibe (substância ativa), a maioria dos pacientes era do sexo masculino (59%) e branco (99%), a mediana da idade era de 56 anos (28% tinham 65 anos ou mais), todos os pacientes apresentavam classificação ECOG de 0 ou 1, e a maioria dos pacientes apresentava doença em estágio M1c (66%). Os desfechos co primários de eficácia do estudo foram sobrevida global (SG) e sobrevida livre de progressão (SLP). Os desfechos-chave secundários incluíram porcentagem de melhor resposta total confirmada (TR) e duração da resposta.
Na análise interina pré-especificada (dados de corte de 30/12/2010), foram observadas melhoras estatística e clinicamente significativas nos desfechos co-primários de sobrevida global (SG) (p < 0,0001) e sobrevida livre de progressão (SLP) (p < 0,0001) (teste log-rank não estratificado). Após recomendação do conselho de monitoramento dos dados de segurança, esses resultados foram divulgados em janeiro de 2011 e o estudo foi modificado para permitir que os pacientes de dacarbazina pudessem passar a receber vemurafenibe (cross-over). Análises de sobrevivência post-hoc foram realizadas posteriormente, conforme descrito na tabela a seguir:
Tabela 1: Sobrevida global em pacientes não tratados previamente com melanoma que apresente mutação positiva para BRAF V600 no estudo com data de corte (N = 338 dacarbazina, N = 337 vemurafenibe)
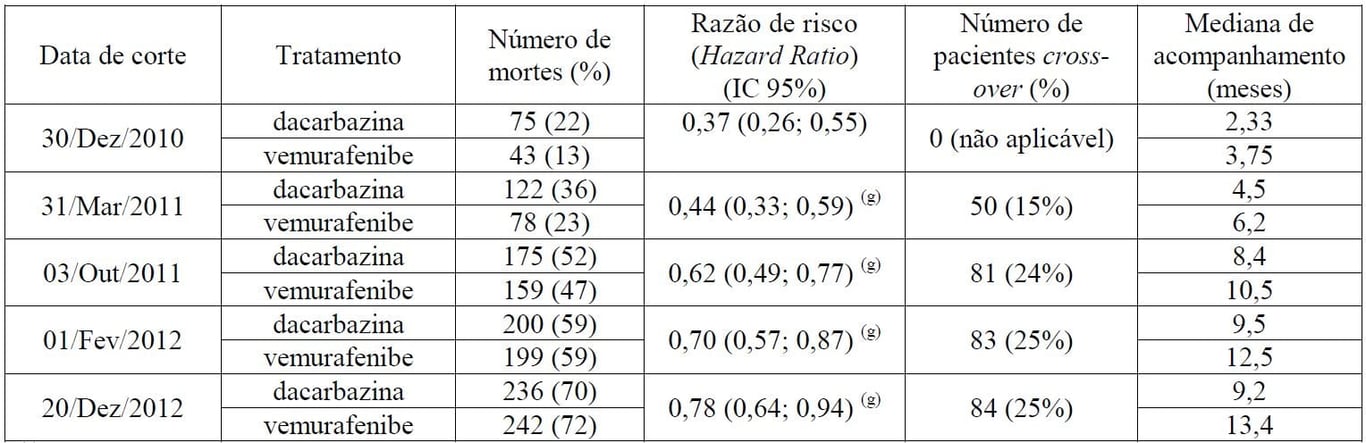
(g) Resultados censurados na época do cross-over.
Resultados não censurados na época do cross-over: 31/Mar/2011: HR (IC 95%) = 0,47 (0,35; 0,62); 03/Out/2011: HR (IC 95%) = 0,67 (0,54; 0,84); 01/Fev/2012: HR (IC 95%) = 0,76 (0,63; 0,93); 20/Dez/2012: HR (IC 95%) = 0,79 (0,66; 0,95).
No momento da atualização de três meses (dados de corte de 31/03/2011), um total de 200 pacientes morreram (78 no braço de Vemurafenibe (substância ativa) e 122 no braço de dacarbazina). O tempo mediano de acompanhamento para a SG no grupo de Vemurafenibe (substância ativa) foi de 6,2 meses (intervalo 0,4 a 13,9 meses) e no grupo de dacarbazina foi de 4,5 meses (intervalo <0,1 a 11,7 meses).
A sobrevida global foi maior no braço de Vemurafenibe (substância ativa), em comparação com o braço de dacarbazina, com razão de risco (Hazard Ratio) de 0,44 (IC 95%: 0,33; 0,59), o que representa redução de 56% do risco de óbito com Vemurafenibe (substância ativa), em comparação com dacarbazina. As estimativas de Kaplan-Meier (K-M) das porcentagens de sobrevida em seis meses foram de 83% (IC 95%: 79%, 87%) para Vemurafenibe (substância ativa) e 63% (IC 95%: 57%, 69%) para dacarbazina. No momento da análise, estimativas de K-M para SG mediana para Vemurafenibe (substância ativa) não foram alcançadas (IC 95%: 9,6; não alcançado) e para dacarbazina foram 7,9 meses (IC 95%: 7,3; 9,6).
Vinte e quatro meses após o último paciente ser randomizado (data de corte dos dados: 20/12/2012)1, foi realizada uma análise post-hoc atualizada da SG. No momento da análise, 478 pacientes tinham morrido (242 no braço de Vemurafenibe (substância ativa) e 236 no braço de dacarbazina). O tempo de acompanhamento mediano no braço de Vemurafenibe (substância ativa) foi de 13,4 meses (variação de 0,4 a 33,3 meses). A estimativa de K-M para SG mediana para Vemurafenibe (substância ativa) foi de 13,6 meses (IC 95%: 12,0; 15,3).
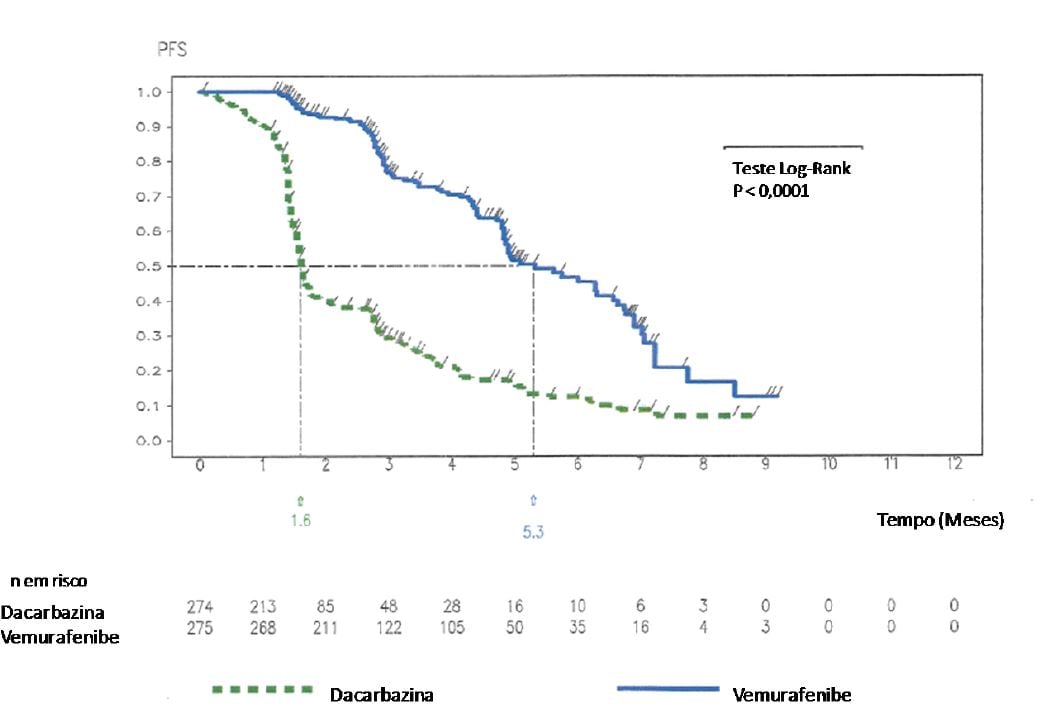
SLP de acordo com avaliação do investigador foi maior com Vemurafenibe (substância ativa), em comparação com dacarbazina, com razão de risco (Hazard Ratio) para progressão ou óbito (SLP) de 0,26 (IC 95%: 0,20; 0,33), o que representa redução de 74% no risco de progressão ou óbito para Vemurafenibe (substância ativa), em comparação com dacarbazina1. A estimativa de Kaplan-Meier das porcentagens de SLP em seis meses foi de 47% (IC 95%: 38%, 55%) para Vemurafenibe (substância ativa) e 12% (IC 95%: 7%, 18%) para dacarbazina. SLP mediana para Vemurafenibe (substância ativa) foi de 5,32 meses (IC 95%: 4,86; 6,57) e para a dacarbazina foi de 1,61 meses (IC 95%: 1,58; 1,74). O desfecho secundário de porcentagem de melhor resposta total confirmada (RP + RC), de acordo com avaliação do investigador, foi significativamente melhor (p < 0,0001) no braço de Vemurafenibe (substância ativa) (48,4%) (IC 95%: 41,6%, 55,2%), em comparação com o braço de dacarbazina (5,5%) (IC 95%: 2,8%, 9,3%). Doença estável, avaliada de acordo com os critérios internacionais de RECIST 1.1, foi observada em 37% dos pacientes tratados com Vemurafenibe (substância ativa) e 24% dos pacientes tratados com dacarbazina.
Melhora em SG, SLP e melhor resposta total confirmada (TR) a favor do tratamento com Vemurafenibe (substância ativa) foram geralmente observadas em todos os subgrupos (idade, sexo, DHL inicial, classificação ECOG, estágio de doença metastática) e regiões geográficas.
Os resultados de eficácia estão resumidos na tabela abaixo e Figuras 1 (SG atualizada) e 2 (SLP).
Tabela 2. Eficácia de Vemurafenibe (substância ativa) em pacientes sem tratamento prévio com melanoma positivo para mutação BRAF V600
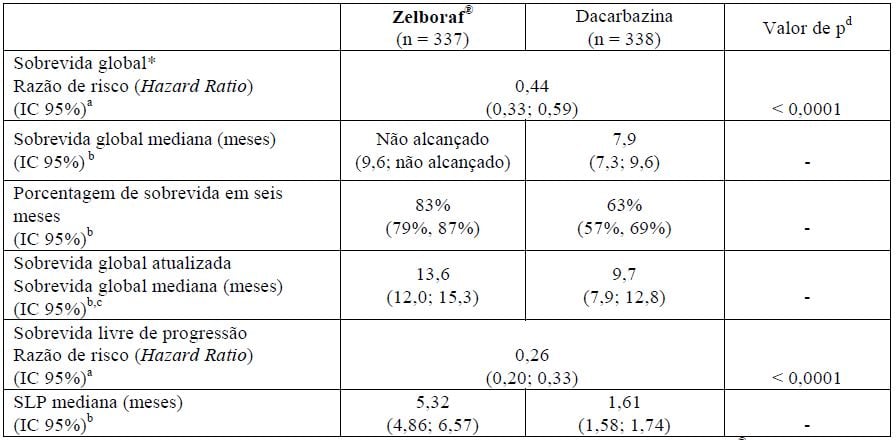
a Razão de risco estimada usando modelo de Cox; uma razão de risco < 1 favorece Vemurafenibe (substância ativa).
b Estimativa de Kaplan-Meier.
c Resultados atualizados (24 meses após a randomização do último paciente).
d Teste log-rank não estratificado.
Figura 1. Curvas de Kaplan-Meier de sobrevida global atualizadas - pacientes sem tratamento prévio (20/12/2012)
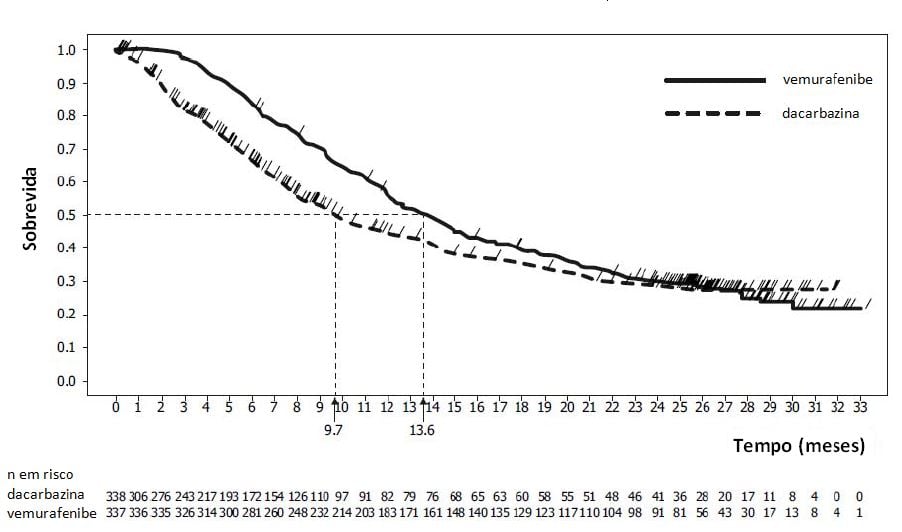
Figura 2. Curvas de Kaplan-Meier de sobrevida livre de progressão - pacientes sem tratamento prévio
A proporção de pacientes com melhora em seu estado clínico geral de acordo com avaliação médica foi maior no grupo de Vemurafenibe (substância ativa) (63,4%) (IC 95%: 57%, 69%) que no grupo de dacarbazina (20,2%) (IC 95%: 15%, 26%).
Pacientes com falha de pelo menos uma terapia sistêmica prévia
Um estudo Fase II de braço único, multicêntrico, multinacional foi conduzido com 132 pacientes com melanoma metastático que tinham recebido pelo menos uma terapia prévia. A idade mediana foi de 52 anos, e 19% dos pacientes tinham mais que 65 anos. A maioria dos pacientes era do sexo masculino (61%), branca (99%) e apresentava doença estágio M1c (61%). Quarenta e nove por cento dos pacientes tinham falhado em duas ou mais terapias prévias.
Com base nos dados com data de corte 01/02/2012, a duração mediana do tratamento foi de 5,7 meses. A duração mediana do acompanhamento foi de 13,4 meses (intervalo de 0,6 a 27,1). O desfecho primário de porcentagem de melhor resposta total confirmada (RP + RC), conforme avaliação de um comitê de revisão independente (CRI), foi de 53% (IC 95%: 44%, 62%). O tempo mediano até a resposta foi de 1,4 meses, com 75% das respostas ocorrendo em torno de 2,6 meses de tratamento. A duração mediana de resposta pelo CRI foi de 7,1 meses (IC 95%: 5,7, 9,8). Foi observada doença estável por RECIST 1.1 em 29% dos pacientes. A sobrevida global mediana foi de 15,9 meses (IC 95%: 11,2; 19,3), e a porcentagem de sobrevida em seis meses foi de 77% (IC 95%: 70%, 85%). A SLP mediana foi de 6,8 meses (IC 95%: 5,5; 7,9) e a porcentagem de SLP em seis meses foi de 54% (IC 95%: 46%, 63%).
Características Farmacológicas
Propriedades farmacodinâmicas
Mecanismo de ação:
Vemurafenibe (substância ativa) é um inibidor de quinase serina-treonina BRAF. Mutações no gene BRAF resultam em ativação constitutiva da proteína BRAF, que pode causar proliferação celular sem associação com fatores de crescimento.
Dados pré-clínicos gerados por testes bioquímicos demonstram que Vemurafenibe (substância ativa) pode inibir potentemente as quinases BRAF com ativação de mutações no códon 600 (veja tabela a seguir).
Tabela 3. Atividade quinase inibitória de Vemurafenibe (substância ativa) contra diferentes BRAF quinases
|
Quinase |
Frequência esperada em melanoma com mutação V600 positiva* |
Concentração inibitória 50 (nM) |
|
BRAFV600E |
87,3% |
10 |
|
BRAFV600K |
7,9% |
7 |
|
BRAFV600R |
1% |
9 |
|
BRAFV600D |
<0,2% |
7 |
|
BRAFV600G |
<0,1% |
8 |
|
BRAFV600M |
0,1% |
7 |
|
BRAFV600A |
<0,1% |
14 |
|
BRAFWT |
NA |
39 |
* Estimado de 16.403 melanomas com mutação BRAF no códon 600 na base de dados pública COSMIC, lançamento 71 (novembro 2014).
Este efeito de inibição foi confirmado na fosforilação ERK e testes de anti-proliferação celular em células disponíveis de melanoma expressando mutação BRAF V600. Em testes de anti-proliferação celular, a concentração inibitória 50 (IC50) contra as linhagens celulares mutadas V600 (linhagens celulares mutadas V600E, V600R, V600D e V600K) variou de 0,016 a 1,131 ?M, ao passo que IC 50 contra linhagens celulares tipo BRAF selvagem foram 12,06 e 14,32 ?M, respectivamente.
Propriedades farmacocinéticas
Os parâmetros farmacocinéticos para Vemurafenibe (substância ativa) foram determinados usando análise não compartimental em um estudo Fase I e um estudo Fase III. As médias de Cmáx, Cmin e AUC (área sob a curva)0-12h foram de, aproximadamente, 62 ?g/mL, 53 ?g/mL e 600 ?g*h/mL. A análise de farmacocinética (PK) populacional usando dados agrupados de 458 pacientes estimou a mediana de Cmáx, Cmin e AUC em estado de equilíbrio dinâmico como sendo de 62 ?g/mL, 59 ?g/mL e 734 ?g*h/mL, respectivamente. A estimativa de razão de acumulação mediana para um esquema duas vezes por dia é de 7,36. Demonstrou-se que a farmacocinética de Vemurafenibe (substância ativa) é proporcional à dose entre 240 e 960 mg, duas vezes por dia, e a análise de farmacocinética populacional também confirmou que a farmacocinética de Vemurafenibe (substância ativa) é linear.
Absorção:
Vemurafenibe (substância ativa) é absorvido com uma mediana de Tmáx de, aproximadamente, quatro horas após uma dose única de 960 mg (quatro comprimidos de 240 mg). Vemurafenibe (substância ativa) apresenta acumulação acentuada depois de administração repetida de 960 mg, duas vezes por dia, com elevada variabilidade entre pacientes. Em um estudo Fase II, a concentração plasmática média de Vemurafenibe (substância ativa), quatro horas depois da administração, aumenta de 3,6 ?g/mL no dia 1 até 49,0 ?g/mL no dia 15 (intervalo de 5,4 a 118 ?g/mL).
Em estado de equilíbrio dinâmico (alcançado no dia 15 em 80% dos pacientes), a exposição média de Vemurafenibe (substância ativa) no plasma é estável (concentrações antes e duas a quatro horas depois da dose matinal), como indicado pela razão de equilíbrio dinâmico, independentemente da redução da dose.
Depois da administração oral, a constante de taxa de absorção para a população de pacientes com melanoma metastático é estimada em 0,19 h-1 (com 101% de variabilidade entre pacientes).
Distribuição:
O volume aparente de distribuição populacional de Vemurafenibe (substância ativa) em pacientes com melanoma metastático é estimado em 91 L (com variabilidade de 64,8% entre os pacientes). In vitro, Vemurafenibe (substância ativa) mostrou-se altamente ligado a proteínas plasmáticas humanas (> 99%).
Metabolismo:
As proporções relativas de Vemurafenibe (substância ativa) e os seus metabólitos foram caracterizados em um estudo de equilíbrio de massa humana com uma dose única de vemurafenibe marcado com C14 administrado oralmente no estado de equilíbrio.
Em média, 95% da dose foi recuperada dentro de 18 dias. A maior parte (94%) nas fezes e < 1% recuperada da urina. Enquanto a CYP3A4 é a enzima primária responsável pelo metabolismo de Vemurafenibe (substância ativa) in vitro, metabolitos conjugados (glucuronidação e glicosilação) foram também identificados em humanos. No entanto, o composto-mãe era o componente predominante no plasma (95%). Embora o metabolismo não pareça resultar em uma quantidade relevante de metabólito no plasma, a importância do metabolismo para a excreção não pode ser excluída.
Início da ação:
No estudo NO25026, o tempo até resposta foi avaliado em pacientes com resposta confirmada. Avaliações tumorais foram realizadas a cada 6 semanas nas primeiras 12 semanas e a cada 9 semanas durante o restante do tratamento. Entre os 106 pacientes do braço de Vemurafenibe (substância ativa) com resposta confirmada, o tempo mediano até resposta foi de 1,45 mês (variação: 1,0 a 5,5). A maioria desses pacientes (75%) respondeu ao tratamento com Vemurafenibe (substância ativa) já na primeira avaliação tumoral pós-basal (1,6 mês).
Eliminação:
A eliminação aparente populacional de Vemurafenibe (substância ativa) em pacientes com melanoma metastático é estimada em 29,3 L/dia (com variabilidade entre pacientes de 31,9%). A mediana de meia-vida de eliminação individual para Vemurafenibe (substância ativa) é de 56,9 horas (o intervalo entre os percentis 5º e 95º é de 29,8 - 119,5 horas).
Farmacocinética em populações especiais:
Idosos
Com base na análise de farmacocinética populacional, a idade não tem efeito estatisticamente significativo sobre a farmacocinética de Vemurafenibe (substância ativa).
Sexo
Na análise de farmacocinética populacional, descobriu-se que o sexo é estatisticamente significativo na explicação da variabilidade entre os pacientes, com uma eliminação aparente 17% maior e um volume aparente de distribuição 48% maior em homens. No entanto, os resultados da análise populacional mostraram que as diferenças em exposição são relativamente pequenas (com uma AUC12 horas e Cmáx em estado de equilíbrio dinâmico estimadas de 792 ?g*h/mL e 67 ?g/mL em mulheres e 696 ?g*h/mL e 63 ?g/mL em homens, respectivamente), indicando que não existe necessidade de ajuste de dose com base no sexo.
Crianças
Nenhum estudo foi conduzido para investigar a farmacocinética de Vemurafenibe (substância ativa) em crianças.
Insuficiência renal
Não foram conduzidos estudos farmacocinéticos de Vemurafenibe (substância ativa) em pacientes com função renal comprometida.
Insuficiência hepática
Com base em dados pré-clínicos e estudo de equilíbrio de massa humana, Vemurafenibe (substância ativa) é eliminado principalmente pelo fígado. Não foi conduzido nenhum estudo farmacocinético de Vemurafenibe (substância ativa) em pacientes com insuficiência hepática.
Cuidados de Armazenamento
Zelboraf deve ser conservado em temperatura ambiente (entre 15 e 30 ºC), na embalagem original, protegido da umidade.
Número de lote e datas de fabricação e validade: Vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Descarte de medicamentos não utilizados e / ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS - 1.0100.0656
Farm. Resp.:
Tatiana Tsiomis Díaz – CRF-RJ n° 6942
Fabricado para
F. Hoffmann-La Roche Ltd, Basileia, Suíça
Por Roche S.p.A., Segrate, Milão, Itália
Zelboraf é comercializado sob licença de Plexxikon Inc., membro do grupo Daiichi Sankyo.
Registrado, importado e distribuído no Brasil por
Produtos Roche Químicos e Farmacêuticos S.A.
Estrada dos Bandeirantes, 2.020
CEP 22775-109 - Rio de Janeiro - RJ
CNPJ: 33.009.945/0023-39
Serviço Gratuito de Informações - 0800 7720 289
www.roche.com.br
Venda sob prescrição médica.
Comparar preços de remédios e medicamentos no CliqueFarma é rápido e simples.
O CliqueFarma, é uma ferramenta para comparativo de preços de produtos farmacêuticos. Não comercializamos, não indicamos, não receitamos, nenhum tipo de medicamento essa função cabe exclusivamente a médicos e farmacêuticos. Não consuma qualquer tipo de medicamento sem consultar seu médico.
SE PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO. PROCURE UM MÉDICO E O FARMACÊUTICO. LEIA A BULA.
Conheça nossos Termos de Uso