- Medicamentos
- Medicamentos de A-Z
- Zavesca 100Mg Com 90 Cápsulas
Comparamos o preço de zavesca 100mg com 90 cápsulas, veja o menor preço
Zavesca 100Mg Com 90 Cápsulas

Menor Preço
R$ 32.700,00
- CATEGORIA: Medicamentos de A-Z
- PRINCÍPIO ATIVO: Miglustate
- FABRICANTE: ACTELION
PARA QUE SERVE?
Para que serve Zavesca é indicado para o tratamento oral da doença de Gaucher do tipo 1 leve a moderada para as quais a terapia de substituição enzimática é considerada inadequada.
R
Referência
7
ofertasMelhores preços a partir de R$ 32.700,00 até R$ 35.947,00
Menor preço
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Onco Express Medicamentos Especiais e Oncológicos
economize
1.15%
R$ 35.532,21
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Para que serve
Zavesca é indicado para o tratamento oral da doença de Gaucher do tipo 1 leve a moderada para as quais a terapia de substituição enzimática é considerada inadequada.
Zavesca é indicado para o tratamento das manifestações neurológicas progressivas em pacientes adultos e pediátricos com doença de Niemann-Pick tipo C.
Como este medicamento funciona?
Zavesca pertence a um grupo de medicamentos que tratam desordens metabólicas. Zavesca inibe a enzima glucosilceramida sintase, responsável pela formação da glucosilceramida também chamada glucocerebrósido. É utilizado para o tratamento de duas condições:
- • Zavesca é usado no tratamento da doença de Gaucher tipo 1 leve a moderada: A doença de Gaucher é uma condição em que há um acúmulo de glucocerebrósido em certas células do sistema imunológico chamadas macrófagos. Isto conduz à deposição da substância nos órgãos do corpo, produzindo, por exemplo, aumento do fígado e baço, alterações no sangue, e doenças ósseas.
- • Zavesca é usado no tratamento de sintomas neurológicos progressivos em Niemann-Pick C: Niemann-Pick C é uma condição em que há um acúmulo de glucocerebrósido nas células cerebrais. Isto leva a distúrbios das funções neurológicas como o movimento dos olhos, equilíbrio, deglutição, memória e convulsões.
Contraindicação
Você não deve utilizar Zavesca:
- • Se você é alérgico (hipersensível) ao miglustate ou a qualquer um dos excipientes da formulação;
- • Se você estiver grávida;
- • Se você estiver amamentando, a menos que recomendado por seus médicos;
- • Se você está pensando em engravidar.
"Este medicamento não pode ser tomado por mulheres grávidas ou que estão pensando em engravidar durante o tratamento."
Como usar
As cápsulas inteiras de Zavesca tem que ser engolidas com água.
Siga a orientação do seu médico, respeitando horários, as doses e a duração do tratamento.
Para a doença de Gaucher tipo 1
A dose inicial recomendada para o tratamento de pacientes com a doença de Gaucher tipo 1 é de 100 mg, três vezes ao dia (manhã, tarde e noite), em intervalos regulares. Se você tem deficiências renais, informe o seu médico.
Para a doença de Niemann- Pick tipo C
Para adultos e adolescentes, a dose habitual é de duas cápsulas (200 mg), três vezes ao dia (manhã, tarde e noite). Isto significa uma dose diária máxima de seis cápsulas (600 mg).
Se você tem menos de 12 anos de idade, o seu médico irá ajustar a dose para a doença de Niemann Pick - tipo C.
Se você tem problema nos rins você pode receber uma dose inicial mais baixa. O seu médico pode reduzir a dose, por exemplo, para uma cápsula (100 mg) uma ou duas vezes ao dia, se você sofre de diarreia ao tomar Zavesca. O seu médico irá dizer quanto tempo o seu tratamento irá durar.
Não interrompa o tratamento sem falar com o seu médico. "Zavesca não deve ser dividido ou mastigado."
Se você tomar mais cápsulas de Zavesca do que o recomendado, consulte imediatamente o seu médico. Zavesca tem sido utilizado em doses dez maiores do que a recomendada em estudos clínicos: isto causou uma redução das células sanguíneas e outras reações adversas descritas nesta bula.
O que devo fazer se eu esquecer de usar Zavesca?
Se você esqueceu qualquer dose, tome a próxima cápsula no horário habitual. Não tome o dobro da dose para compensar uma dose que você esqueceu.
Se você parar de tomar Zavesca: Não pare de tomar Zavesca sem falar com o seu médico.
Em caso de dúvidas, ligue para o seu médico ou farmacêutico.
Precauções
Zavesca não deve ser utilizado por mulheres grávidas ou mulheres que estão tentando engravidar. Os homens tratados com Zavesca devem utilizar métodos contraceptivos seguros durante o tratamento, incluindo três meses após a interrupção do tratamento.
Você pode pedir para seu médico a avaliação antes e durante o tratamento com Zavesca dos nervos dos braços e pernas, os níveis de vitamina B12, monitoramento do crescimento em crianças e adolescentes portadores de Niemann-Pick C, monitoramento da contagem de plaquetas em pacientes com Niemann-Pick C. A razão para solicitar estes testes é que alguns pacientes tiveram dormência ou formigamento nas mãos ou pernas e redução de peso durante o tratamento com Zavesca. Estes testes irão ajudar o médico a avaliar se esses efeitos são devidos à doença ou outras condições existentes, ou tratar reações adversas a medicamentos.
Se você tiver diarreia, o médico pode recomendar uma mudança na dieta para reduzir a ingestão de lactose e de carboidratos, ou não tomar Zavesca com alimentos ou reduzir temporariamente a dose. Em alguns casos, o médico pode prescrever um anti-diarreico tal como a loperamida. Se você não melhorar, ou apresentar dor abdominal, consulte o seu médico. O seu médico poderá investigar mais.
Zavesca pode causar tonturas. Se você sentir tonturas, não dirigir ou operar máquinas durante o tratamento com Zavesca.
Antes de utilizar Zavesca, informe o seu médico se:
- • Você está grávida ou pretende engravidar;
- • Você está amamentando;
- • Você não está utilizando um método contraceptivo confiável;
- • Você apresentar problemas renais ou hepáticos;
- • Você está tomando ou tomou recentemente outros medicamentos.
"Os eventos adversos devem ser comunicados ao seu médico."
"Informe ao seu médico ou dentista se você estiver usando qualquer outro medicamento." Cerezyme administrado em conjunto com Zavesca pode diminuir a exposição do seu corpo ao Zavesca.
"Não use medicamento sem informar ao seu médico. Pode ser perigoso para a sua saúde."
Reações Adversas
Como todos os medicamentos, Zavesca pode causar reações adversas, embora nem todas as pessoas apresentem.
Se você perder peso no início do tratamento com Zavesca, não se preocupe. Os pacientes geralmente não conseguem perder peso durante o tratamento.
Se você tiver um tremor leve, mais comumente nas mãos, informe o seu médico assim que possível. Tipicamente, o tremor desaparece sem a necessidade de interrupção do tratamento. Pode ser que seu médico necessite reduzir a dose ou interromper Zavesca para parar o tremor.
Os efeitos indesejáveis são listados por frequência, usando a seguinte convenção: muito comum (? 1/10), comum (1/100 ? x < 1/10), pouco comum (> 1/10.000, ? 1/1.000), muito raros (? 1/10.000). As reações adversas estão organizadas de acordo com a frequência (das mais frequente para as menos frequentes).
Distúrbios Sanguíneos e do Sistema Linfático:
- Comum: trombocitopenia
Distúrbios do Metabolismo e Nutrição:
- Muito Comum: perda de peso, diminuição do apetite
Distúrbios Psiquiátricos
- Comum: depressão, insônia, diminuição da libido
Distúrbios do Sistema Nervoso
- Muito Comum: tremor
- Comum: neuropatia periférica, dor de cabeça, parestesia, tontura, coordenação anormal, hipoestesia, amnésia
Distúrbios Gastrointestinais
- Muito Comum: diarreia, flatulência, dor abdominal
- Comum: náusea, vômito, distenção/desconforto abdominal, Constipação e dispepsia
Distúrbios músculo-esqueléticos e de tecidos conjuntivos
- Comum: espasmos musculares, fraqueza muscular
Distúrbios Gerais e reações no local de administração
- Comum: fadiga, astenia, calafrios, mal estar
Investigações
- Comum: condução nervosa (anormal)
As reações adversas mais comuns são: perda de peso, tremores, diarreia, flatulência (gases intestinais) e dor abdominal (estômago). Entre as reações adversas comuns estão incluídas: anorexia (perda de apetite), diminuição do apetite, dor de cabeça, tonturas, neuropatia periférica, parestesia (incluindo dormência e alterações da sensibilidade), coordenação anormal, hipoestesia (diminuição da sensibilidade ao tato), dispepsia, azia, náuseas, constipação e vômitos, inchaço ou desconforto no abdômen (barriga) e trombocitopenia (redução nos níveis de plaquetas). Os sintomas neurológicos e trombocitopenia podem ser devidos à própria doença. Outras reações adversas possíveis incluem: espasmos musculares (ou fraqueza), fadiga, dificuldade em dormir, e diminuição da libido.
A maior parte dos pacientes apresentou um ou mais destes efeitos adversos, tipicamente, no início do tratamento e a intervalos regulares durante o tratamento. A maioria dos casos são leves e desaparecem muito rápido. Se algum destes efeitos colaterais causar problemas, consulte o seu médico. Ele ou ela pode reduzir a dose de Zavesca ou recomendar um medicamento para ajudar no controle das reações adversas.
Alguns pacientes apresentaram alterações na sensibilidade e formigamento nas mãos e pés. Essas reações podem ser sinais de neuropatia periférica devido aos efeitos adversos de Zavesca ou pode surgir a partir de condições existentes. O seu médico irá realizar alguns testes antes e durante o tratamento com Zavesca para uma melhor avaliação. Se você sentir qualquer uma destas reações adversas, informe o seu médico assim que possível.
Se algum dos efeitos colaterais se agravar ou se você detectar quaisquer efeitos indesejáveis não mencionados nesta bula, informe o seu médico.
"Atenção: este é um medicamento novo e, embora estudos clínicos tenham demonstrado eficácia e segurança aceitáveis , podem ocorrer efeitos indesejáveis e desconhecidos. Neste caso, informe seu médico."
Informe o seu médico, dentista ou farmacêutico quaisquer reações indesejáveis pelo uso do medicamento. Informe também a empresa através do seu serviço ao cliente.
População Especial
Gravidez e amamentação
Você não deve tomar Zavesca se você está grávida ou pretende engravidar. Seu médico pode dar-lhe informações adicionais. Você deve usar um método contraceptivo confiável durante o tratamento com Zavesca. Não amamente enquanto estiver tomando Zavesca.
Consulte o seu médico antes de tomar qualquer medicamento.
Alterações laboratoriais
Não foi encontrada interação de miglustate em testes laboratoriais.
Composição
Zavesca
Miglustate
Forma Farmacêutica
Cápsula:
100 mg, embalagem com 5 blisters com 18 cápsulas em cada blister.
Para administração oral.
USO ADULTO E PEDIÁTRICO
Composição
Cada cápsula de Zavesca contém: 100 mg de miglustate.
Excipientes: Amido glicolato de sódio, povidona (K 30), e estearato de magnésio.
Superdosagem
Se tomar mais cápsulas do que lhe prescrito, consulte o seu médico imediatamente. Zavesca tem sido utilizado em estudos clínicos em doses dez vezes maiores do que a dose recomendada: Isto causou diminuição nas células brancas do sangue e outros efeitos colaterais similares.
Não há experiência de sobredosagem com Zavesca em pacientes. Zavesca foi utilizado em ensaios clínicos em doses superiores a 10 vezes a dose recomendada. A redução na contagem de células brancas foi observada nestas doses. Todas as outras reações adversas foram semelhantes às descritas no item 8.
Em caso de sobredosagem, o médico deve ser chamado ou um hospital de emergência deve ser procurado imediatamente.
Todos os medicamentos devem ser mantidos fora do alcance das crianças.
Em caso de intoxicação ligue 0800 722 6001, se você precisar de mais orientações sobre como proceder.
Interação Medicamentosa
Dados limitados sugerem que a coadministração deste medicamento e imiglucerase pode resultar em diminuição da exposição a miglustate (redução aproximada de 22 % na Cmáx e de 14% na AUC foram observadas em um pequeno estudo de grupo paralelo). Este estudo também indicou que miglustate tem limitado ou nenhum efeito sobre a farmacocinética de imiglucerase.
Alterações laboratoriais
Não foi encontrada interação deste medicamento em testes laboratoriais.
Interação Alimentícia
Miglustate pode ser tomado com ou sem alimentos.
Ação da Substância
Resultados de eficácia
A segurança e a eficácia do Miglustate (substância ativa) na doença de Gaucher tipo 1 foram investigadas em três estudos abertos, não comparativos e um estudo aberto, randomizado, controlado com terapia de reposição enzimática (reposição enzimática com imiglucerase).
Parâmetros de eficácia incluíram a avaliação dos volumes do fígado e do baço, a concentração de hemoglobina e contagem de plaquetas. Estudos abertos, não controlados, em monoterapia
No Estudo OGT 918-001, Miglustate (substância ativa) foi administrado na dose de 100 mg, três vezes ao dia, para pacientes incapazes ou não dispostos a receber a terapia de reposição enzimática, ou que não tinham tomado a terapia de reposição enzimática nos últimos 3 meses. Vinte e oito pacientes com doença de Gaucher tipo I leve a moderada foram incluídos neste estudo de 12 meses, 22 pacientes completaram o estudo.
Em 12 meses, houve um percentual médio de redução significativo a partir do basal no volume do fígado de 12,1% e no volume do baço de 19,0%, um aumento do basal na concentração média de hemoglobina de 0,26 g/dL e um aumento na contagem média das plaquetas de 8,29 x 109/L. Dezoito pacientes continuaram a receber Miglustate (substância ativa) em um protocolo opcional de extensão de tratamento.
O benefício clínico foi avaliado em 24 e 36 meses, em 13 pacientes. Após 3 anos de tratamento contínuo com Miglustate (substância ativa), as reduções médias nos volumes de fígado e baço foram significativas em 17.5 % e 29,6 %, respectivamente. Houve aumentos médios significativos de 22,2 x 109/L na contagem de plaquetas e de 0,95 g/dL na concentração de hemoglobina.
No Estudo OGT 918-003, Miglustate (substância ativa) foi administrado na dose de 50 mg, três vezes ao dia, durante 6 meses a 18 pacientes adultos com doença de Gaucher tipo 1 leve a moderada que não podiam ou não queriam receber terapia de reposição enzimática, ou que não tinham tomado a terapia de reposição enzimática nos últimos 3 meses.
Dezessete pacientes completaram o estudo. Após 6 meses de tratamento, os resultados mostraram percentual médio de redução significativo a partir do basal no volume do fígado de 6% e no volume do baço de 5%.
Houve uma diminuição absoluta média não significativa a partir do basal na concentração de hemoglobina de 0,13 g/dL e um aumento não significativo a partir do basal na contagem de plaquetas de 5 x 109/L na dose utilizada neste estudo (50 mg, três vezes ao dia).
Dezesseis pacientes continuaram a receber Miglustate (substância ativa) em um protocolo opcional de extensão de 6 meses de tratamento. Após 12 meses de tratamento contínuo com Miglustate (substância ativa), houve uma redução média do volume do baço de 10 %, enquanto que a redução média do volume do fígado ficou em 6%.
Os resultados do Estudo OGT 918-001 e do Estudo OGT 918-003 indicam uma maior eficácia associada a Miglustate (substância ativa) 100 mg, três vezes ao dia, em comparação com 50 mg, três vezes ao dia.
No Estudo OGT 918-005, Miglustate (substância ativa) foi administrado na dose de 100 mg, três vezes ao dia, para 10 pacientes com doença de Gaucher tipo 1 leve a moderada que não podiam ou não queriam receber a terapia de reposição enzimática, ou que não tinham tomado a terapia de reposição enzimática nos 3 meses anteriores.
Sete pacientes completaram a fase central do estudo de 12 meses e o período opcional de extensão do estudo de até 24 meses. As reduções médias percentuais a partir do basal no volume de fígado foram avaliadas nos Meses 6, 12, 18 e 24. Estas reduções foram significativas ao mês 6 (8,4%) e Mês 18 (15,1%). Reduções no volume do fígado no Mês 12 e Mês 24 indicaram um efeito consistente do tratamento.
As reduções absolutas médias e percentuais a partir do basal no volume do baço foram avaliadas nos Meses 6, 12, 18 e 24. Reduções médias significativas foram observadas no Mês 6 (19,0%) e Mês 18 (24,3%). As concentrações médias de hemoglobina, que eram normais no basal, mantiveram-se estáveis durante o curso do estudo. Houve um aumento médio significativo na contagem de plaquetas no Mês 12 (13,9 x 109/L). O envolvimento ósseo avaliado por MRI/BMD manteve-se estável durante os 24 meses do estudo.
Estudo aberto, controlado com ativo
O Estudo OGT 918-004 foi um estudo aberto, randomizado, controlado por ativo em 36 pacientes com a doença de Gaucher tipo 1 que receberam um mínimo de dois anos de tratamento com TRE com terapia de reposição enzimática com imiglucerase antes do início do estudo clínico e que estavam em uma dose estável desde pelo menos seis meses.
O estudo teve duas fases: uma fase comparativa de seis meses, e uma fase de longa duração, permitindo o tratamento com Miglustate (substância ativa) como monoterapia por até 24 meses após a retirada da TRE.
Na fase inicial de 6 meses, 36 pacientes foram randomizados 1:1:1 para um dos três grupos de tratamento, como se segue:
- Continuação da monoterapia com imiglucerase (na dose usual utilizada pelo paciente);
- Combinação de imiglucerase (na dose usual utilizada pelo paciente) com Miglustate (substância ativa) (100 mg, três vezes ao dia);
- Mudar para monoterapia com Miglustate (substância ativa) (100 mg, três vezes ao dia).
Trinta e três pacientes completaram a fase inicial de 6 meses do estudo, dos quais 29 foram incluídos na fase de extensão inicial de 6 meses. Vinte e oito desses 29 pacientes, posteriormente, entraram em uma segunda fase de extensão por mais 12 meses recebendo Miglustate (substância ativa) isoladamente.
Na fase inicial de 6 meses do estudo, as alterações médias nos volumes do fígado e baço e nos parâmetros hematológicos foram pequenas e clinicamente não significativas em todos os três grupos de tratamento.
Uma análise retrospectiva dos dados individuais do paciente foi realizada para os 29 pacientes que contribuíram com dados sobre Miglustate (substância ativa) em monoterapia de manutenção para avaliar os desfechos em relação à estabilidade da doença no momento da retirada da TRE. Os critérios foram baseados em relatórios de consenso publicado para TRE e discussão de especialistas, como se segue:
Critérios para a estabilização da doença na retirada da TRE (todos dos seguintes): - Tratamento com TRE por pelo menos três anosDoença estável nos últimos dois anos:
Volumes do fígado e baço:
Últimos 3 valores dentro de 10% da média desses 3 valores.
Doença óssea:
Sem sinais e sintomas de doença óssea grave (crises ósseas, necrose avascular, fratura patológica) nos dois anos anteriores. Ausência de dor óssea atual exigindo medicamento para a dor crônica ou que interfere com a vida cotidiana.
Hemoglobina:
Controlada e estável acima de 11 g/dL.
Plaquetas:
Controladas e estáveis, pelo menos, 100 x 109/L.
Quitotriosidase:
Últimos 3 valores dentro de 20% da média desses 3 valores.
Critérios para a eficácia do Miglustate (substância ativa) em monoterapia de manutenção:
Foram definidos critérios de sinal de potencial falha para manter a doença estável durante a monoterapia com Miglustate (substância ativa) (pelo menos uma das seguintes). Tais sinais devem solicitar novas investigações e revisão do tratamento:
- Aumento do basal no volume do fígado de pelo menos 10%.
- Aumento do basal no volume do baço de pelo menos 10%.
- Redução do basal de Hb ? 1 g/dL desde que a concentração resultante seja menor do que o intervalo do limite inferior do normal.
Redução do basal na contagem de plaquetas:
- Abaixo de 120x109/L, se o valor inicial estiver dentro da faixa normal de ? 30x109/L, se o valor basal foi menor do que o limite inferior do normal.
- Qualquer redução que leva a um valor < 50x109/L ou associada a hematomas ou hemorragias clinicamente significativas - Quaisquer outros sinais clínicos significativos e sintomas sugestivos de agravamento da doença.
Quinze dos 29 pacientes preencheram os critérios acima para a estabilização da doença na retirada da TRE. A exposição média ao Miglustate (substância ativa) em monoterapia foi de 19 meses. A manutenção do estado de doença no basal foi atingida com Miglustate (substância ativa) em monoterapia na maioria dos pacientes (11/15).
Os outros quatro pacientes desenvolveram um ou mais sinais de potencial de falha para manter a doença estável. Independentemente do grau de estabilização da doença durante o período de retirada da TRE, nenhum dos pacientes desenvolveu uma deterioração rápida após a troca para a terapia com Miglustate (substância ativa).
Os dados para apoiar a segurança e a eficácia do Miglustate (substância ativa) na doença de Niemann- Pick tipo C vem de um estudo clínico aberto e prospectivo e um estudo retrospectivo. O estudo clínico incluiu 29 pacientes adultos e juvenis em um período controlado de 12 meses, seguido por terapia de extensão para uma duração total média de 3,9 anos e até 5,6 anos.
Além disso, 12 pacientes pediátricos foram incluídos em um sub-estudo não controlado por uma duração geral média de 3,1 anos e até 4,4 anos. Entre os 41 pacientes incluídos no estudo, 14 pacientes foram tratados com Miglustate (substância ativa) por mais de 3 anos. A pesquisa incluiu uma série de casos de 66 pacientes tratados com este medicamento fora do estudo clínico para uma duração média de 1,5 anos.
Em ambos os conjuntos de dados foram incluídos pacientes pediátricos, adolescentes e adultos com idade entre 1 ano a 43 anos. A dose habitual de Miglustate (substância ativa) pacientes adultos é de 200 mg, três vezes ao dia, e foi ajustada de acordo com a área de superfície corporal em pacientes pediátricos.
O desfecho primário avaliou a mudança do basal na velocidade de movimento sacádico horizontal olho (HSEM), expressa em HSEM - ?. Em pacientes tratados com este medicamento uma melhoria significativa (redução da HSEM - ?) em comparação com o valor basal foi observada contra uma deterioração no grupo não tratado. Pacientes pediátricos tratados com este medicamento também mostraram melhora a partir do basal.
Tabela 1 Mudança a partir do basal no HSEM - ? por até 12 meses, Estudo OGT 918-007
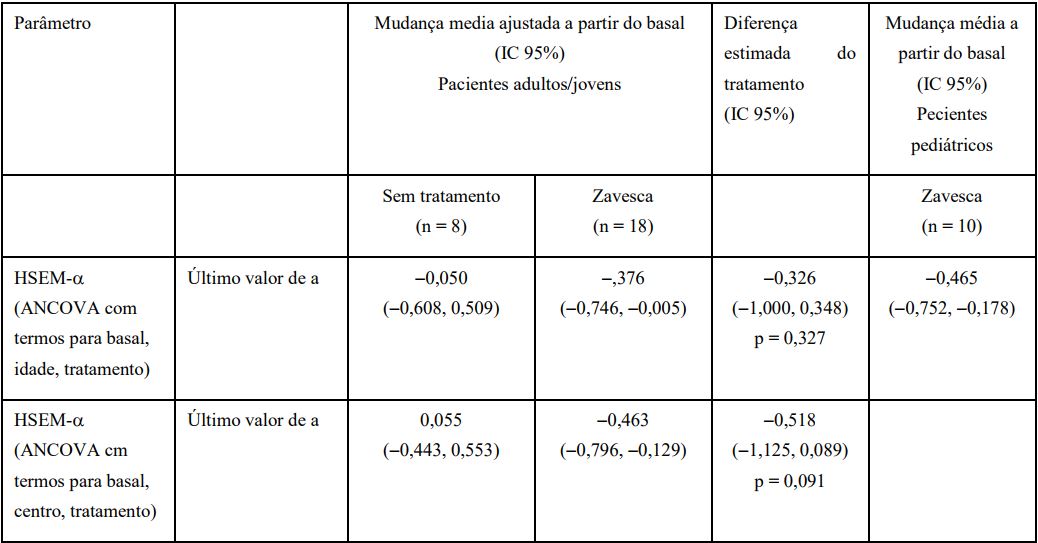 a Último valor é o último valor pós-basal até ao Mês 12. O aumento do basal indica piora.
a Último valor é o último valor pós-basal até ao Mês 12. O aumento do basal indica piora.
IC = intervalo de confiança; HSEM = movimento sacádico horizontal do olho.
A função deglutição foi avaliada numa escala de classificação, avaliando a capacidade do paciente em engolir água e comida de diferentes consistências. A melhor manutenção da função de deglutição foi observada com o tratamento com este medicamento versus nenhum tratamento (risco relativo de qualquer deterioração até Mês 12: 0,4 [IC 95% 0,13, 1,22, p = 0,17]). No geral, cerca de 80 % dos pacientes adultos / juvenil e infantil mantiveram pelo menos deglutição estável em 24 meses de tratamento com este medicamento.
A deficiência motora foi avaliada com o Índice de Hauser Standard Ambulation (SAI). A melhor manutenção da função deambulação (menor deterioração a partir do basal na média SAI) foi observada com o tratamento com este medicamento versus nenhum tratamento durante o estudo controlado de 12 meses em pacientes adultos/juvenis (este medicamento: 0,087 [IC 95% - 0,287, 0,461 ], nenhum tratamento: 0,802 [IC 95% 0,220, 1,385], efeito do tratamento [ANCOVA com os termos basal, centro, grupo de tratamento]: -0,715 [IC 95% - 1,438, 0,007, p = 0,052]). Após 2 anos de tratamento com este medicamento, dois terços dos pacientes adultos/juvenis e infantis mantiveram pelo menos a capacidade de deambulação estável.
A avaliação da capacidade cognitiva, medida através da mudança do basal pelo escore de Folstein Mini-Mental Status Examination (MMSE) em pacientes adultos/juvenis, também mostrou uma diferença em favor deste medicamento durante a fase de 12 meses de estudo controlado OGT 918-007 (este medicamento: 1,219 [ IC 95% - 0,060, 2,498], nenhum tratamento: - 0,352 [IC 95% -2.213, 1,510], efeito do tratamento [ANCOVA com os termos basal, centro, grupo de tratamento]: -1,571 [IC 95% -0,692, 3,834, p = 0,165]).
Em geral, os dados mostraram que o tratamento com Miglustate (substância ativa) pode reduzir a progressão de sintomas neurológicos clinicamente relevantes em pacientes com doença de Niemann-Pick C.
Dados adicionais para apoiar a eficácia do Miglustate (substância ativa) vêm de uma pesquisa retrospectiva [D- 08.406] que compreende uma série de casos de 66 pacientes com doença de Niemann Pick- tipo C tratados com este medicamento para uma duração média de 1,5 anos, após uma observação média de pré-tratamento de 3,1 anos. Este conjunto de dados também incluiu pacientes pediátricos, juvenis e adultos, com faixa etária de 1 a 43 anos.
A progressão da doença foi avaliada dentro dos domínios funcionais de deglutição, deambulação, manipulação (dismetria/distonia), função/articulação da linguagem e da deficiência em geral de acordo com uma escala de incapacidade publicada para a doença de Niemann Pick- tipo C.
Entre domínios funcionais e para a deficiência em geral, este medicamento foi associado com reduções clinicamente relevantes na taxa de progressão anual, em comparação com o pré-tratamento.
O benefício do tratamento com Miglustate (substância ativa) em manifestações neurológicas em pacientes portadores de Niemann-Pick C deve ser avaliado regularmente, como por exemplo, a cada 6 meses: a continuação do tratamento deve ser reavaliada após, pelo menos, 1 ano de tratamento com Miglustate (substância ativa).
Dados Pré-Clínicos de Segurança
Os principais efeitos comuns a todas as espécies foram diarreia e perda de peso, e, em doses mais altas, danos na mucosa gastrointestinal (erosões e ulceração). Outros efeitos observados em animais em doses que resultam em níveis de exposição moderadamente mais altos do que o nível de exposição clínico foram: alterações dos órgãos linfoides em todas as espécies testadas, alterações das transaminases, vacuolização da tireoide e do pâncreas, cataratas, nefropatia e alterações miocárdicas em ratos. Estes resultados foram considerados secundários à debilitação.
A administração de Miglustate (substância ativa) a ratos Sprague Dawley do sexo masculino e do sexo feminino por sonda oral durante 2 anos em níveis de dosagem de 30, 60 e 180 mg/kg/dia resultou em um aumento da incidência hiperplasia das células (células de Leydig) e adenomas intersticiais testiculares em ratos machos com todas as doses.
A exposição sistêmica com a dose mais baixa foi comparável à observada em seres humanos (com base em AUC0-?) na dose recomendada para seres humanos. A ausência de efeitos observáveis (NOEL) não foi estabelecida e o efeito não foi dependente da dose. Não houve aumento relacionado com o fármaco na incidência de tumores em ratos machos ou fêmeas em qualquer outro órgão.
O mecanismo destes achados em ratos ainda não é conhecido. Os tumores de células intersticiais testiculares com compostos não genotóxicos são geralmente considerados de pouca relevância para os seres humanos.
A administração de Miglustate (substância ativa) para camundongos CD1 machos e fêmeas por sonda oral em doses de 210, 420 e 840/500 mg/kg/dia (redução da dose após meio ano) durante 2 anos, resultou em um aumento da incidência de lesões inflamatórias e hiperplásicas no intestino grosso em ambos os sexos.
Com base em mg/kg/dia, e corrigida para diferenças na excreção fecal, as doses corresponderam a 16, 32 e 65/38 vezes a dose recomendada para o homem. Houve descobertas ocasionais de lesões neoplásicas, principalmente na dose mais elevada de 840/500 mg/kg/dia. Os carcinomas de intestino grosso ocorreram ocasionalmente em todas as doses, com um aumento estatisticamente significativo no grupo de dose elevada.
A relevância destas descobertas para os seres humanos não pode ser excluída. Não houve aumento relacionado com o fármaco na incidência de tumores em qualquer outro órgão.
Miglustate (substância ativa) não mostrou nenhum potencial para efeitos mutagênicos ou clastogênicos na bateria padrão de testes de genotoxicidade.
Os estudos de toxicidade de doses repetidas em ratos mostraram efeitos sobre o epitélio seminífero dos testículos. Outros estudos revelaram alterações nos parâmetros de esperma (motilidade e morfologia) consistente com uma redução da fertilidade observada. Estes efeitos ocorreram em níveis de exposição semelhantes aos dos pacientes, mas mostraram reversibilidade.
Miglustate (substância ativa) afetou a sobrevida embrionária/fetal em ratos e coelhos, foi relatada distocia, perdas pós-implantação aumentaram, e um aumento da incidência de anomalias vasculares ocorreu em coelhos. Estes efeitos podem ser, em parte, relacionados à toxicidade materna.
Alterações na lactação foram observados em ratos fêmeas em um estudo de 1 ano. O mecanismo para este efeito é desconhecido.
Características Farmacológicas
Propriedades farmacodinâmicas
Doença de Gaucher tipo 1
A doença de Gaucher é uma doença metabólica hereditária causada por uma falha na degradação da glicosilceramida resultando em depósito lisossômico deste material e patologia generalizada. Miglustate (substância ativa) é um inibidor da glucosilceramida sintase, a enzima responsável pelo primeiro passo na síntese da maioria dos glicolípidos. Estudos in vitro e in vivo demonstraram que o Miglustate (substância ativa) pode reduzir a síntese de glucosilceramida. Esta ação inibitória constitui o fundamento lógico para a terapia de redução de substrato na doença de Gaucher.
O estudo piloto de Miglustate (substância ativa) foi conduzido em pacientes incapazes ou não dispostos a receber a terapia de reposição enzimática (TRE). Razões para não receber a TRE incluíram a carga de infusões intravenosas e dificuldades no acesso venoso. Vinte e oito pacientes com doença de Gaucher tipo 1 leve a moderada foram incluídos neste estudo não comparativo de 12 meses, e 22 pacientes completaram o estudo.
No Mês 12, houve uma redução média do volume do baço de 19,0%. Um aumento significativo na concentração de hemoglobina de 0.26g/dL (5,7%) e um aumento significativo da contagem de plaquetas de 8,29 x 109/L (16,0%) foram observados. Dezoito pacientes, em seguida, continuaram a receber este medicamento sob um protocolo opcional de extensão de tratamento.
O benefício clínico foi avaliado em 24 e 36 meses, em 13 pacientes. Após 3 anos de tratamento contínuo com Miglustate (substância ativa), as reduções médias volume do fígado e do baço foram de 17,5% (-19,9 para -15,1, p < 0,001) e 29,6% (-34,1 para -25,2, p < 0,001), respectivamente. Houve um aumento significativo de 22,2 x 109/L (34,3%) na contagem de plaquetas e um aumento significativo de 0.95g/dL (12,9%) da concentração de hemoglobina.
Um segundo estudo aberto, randomizado e controlado com 36 pacientes que receberam um mínimo de 2 anos de tratamento com TRE em três grupos de tratamento: continuação com Cerezyme*, Cerezyme* em combinação com Miglustate (substância ativa) ou mudança para Miglustate (substância ativa).
Este estudo foi realizado durante um período de seis meses de comparação randomizada seguido por extensão de 18 meses em que todos os pacientes receberam Miglustate (substância ativa).
Em pacientes que mudaram para Miglustate (substância ativa), os volumes do fígado e do baço e os níveis de hemoglobina mantiveram-se inalterados. Em alguns pacientes, houve redução do número de plaquetas e aumento da atividade da quitotriosidase, indicando que Miglustate (substância ativa) em monoterapia não pode manter o mesmo controle da atividade da doença em todos os pacientes. Vinte e nove pacientes continuaram no período de extensão.
Não houve alteração nos resultados depois de 18 e 24 meses, em comparação com 6 meses de Miglustate (substância ativa) em monoterapia (20 pacientes e 6, respectivamente). Nenhum paciente apresentou rápida deterioração da doença de Gaucher tipo 1 após a mudança para Miglustate (substância ativa) em monoterapia.
O esquema terapêutico de Miglustate (substância ativa) empregada em ambos os estudos clínicos descritos anteriormente foi de 100 mg, três vezes por dia. Um estudo adicional de monoterapia foi realizado em 18 pacientes com dose diária total de 150 mg. Os resultados indicaram uma eficácia reduzida, em comparação com a dose diária total de 300 mg.
Um estudo aberto, não comparativo, de 2 anos [Estudo OGT 918-011] incluiu 42 pacientes com doença de Gaucher tipo 1, que haviam recebido um mínimo de 3 anos de TRE e que preenchiam os critérios de doença estável por pelo menos 2 anos. Os pacientes foram transferidos para monoterapia com Miglustate (substância ativa) 100 mg, três ao dia. O volume do fígado (variável primária de eficácia) foi inalterada desde o basal até o fim do tratamento.
Seis pacientes tiveram o tratamento com Miglustate (substância ativa) interrompido prematuramente devido ao potencial agravamento da doença, como definido no estudo. Vinte e um pacientes completaram 24 meses de tratamento com Miglustate (substância ativa). Destes, 18 pacientes estavam no basal dentro dos objetivos terapêuticos estabelecidos para o volume do fígado e baço, níveis de hemoglobina e plaquetas, e 16 pacientes permaneceram dentro de todos esses objetivos terapêuticos no mês 24.
Manifestações ósseas da doença de Gaucher tipo 1, foram avaliadas em 3 estudos clínicos abertos em pacientes tratados com Miglustate (substância ativa), 100 mg, três vezes ao dia, por até 2 anos (n = 72). Em uma análise conjunta, a média da densidade mineral óssea no escore Z na coluna lombar e colo do fêmur aumentou mais de 0,1 unidade a partir do basal em 27 (57%) e 28 (65%) dos pacientes que apresentaram medidas longitudinais de densidade óssea. Não houve eventos de crise óssea, necrose avascular ou fratura durante o período de tratamento.
Doença de Niemann-Pick Tipo C
A doença de Niemann-Pick tipo C é uma doença neurodegenerativa rara, invariável, progressiva e, eventualmente, fatal caracterizada tráfego prejudicado de lipídios intracelulares. As manifestações neurológicas são consideradas secundárias ao acúmulo anormal de células glicoespingoneuronal e glial.
Miglustate (substância ativa) mostrou eficácia em modelos animais relevantes da doença de Niemann- Pick C. Miglustate (substância ativa) atravessa a barreira hematoencefálica.
A doença de Niemann-Pick C geralmente começa na infância ou na fase juvenil e é caracterizada pelo desenvolvimento progressivo de ataxia, retardo no crescimento e na prevenção dos movimentos oculares sacádicos que levam à paralisia supranuclear fixa com perturbação visual, disfagia, disartria, convulsões e distonia.
Propriedades farmacocinéticas
Os parâmetros farmacocinéticos de Miglustate (substância ativa) foram avaliados em indivíduos saudáveis, em um pequeno número de pacientes com doença de Gaucher tipo 1, doença de Fabry, pacientes infectados pelo HIV, em adultos, adolescentes e crianças com doença de Niemann- Pick tipo C ou doença de Gaucher tipo 3.
A cinética de Miglustate (substância ativa) parece ser a dose linear e independente do tempo. Em indivíduos saudáveis, Miglustate (substância ativa) é rapidamente absorvido. As concentrações plasmáticas máximas são atingidas cerca de 2 horas após a dose. A biodisponibilidade absoluta não foi determinada. A administração concomitante de alimentos diminui a taxa de absorção (Cmáx diminuiu 36 % e tmáx teve um atraso de 2 horas), mas não há nenhum efeito estatisticamente significativo sobre a extensão da absorção de Miglustate (substância ativa) (AUC diminuiu 14%).
O volume aparente de distribuição é de 83 L. Miglustate (substância ativa) não se liga às proteínas plasmáticas. Miglustate (substância ativa) é eliminado principalmente por excreção renal, com recuperação do fármaco inalterado na urina em 70-80 % da dose.
A biotransformação resulta em uma série de metabolitos que são excretados através da urina e das fezes. A principal via de eliminação é através da urina, com uma recuperação média de 82,8 % da dose administrada. A excreção fecal dá uma recuperação média de 11,9% da dose administrada.O clearance oral aparente (CL/F) é de 230 ± 39 mL/min. A meia-vida média é de 6-7 horas.
Após a administração de uma dose única de 100 mg de 14C Miglustate (substância ativa) a voluntários saudáveis , 83% da radioatividade foi recuperada na urina e 12 % nas fezes. Foram identificados vários metabólitos na urina e nas fezes. O metabólito mais abundante na urina foi Miglustate (substância ativa) glucuronido representando 5 % da dose. A meia-vida da radioatividade no plasma foi de 150 h, sugerindo a presença de um ou mais metabólitos de meia-vida muito longa.
A recuperação para este metabólito não foi identificada, mas pode acumular-se e atingir concentrações superiores a aquelas do Miglustate (substância ativa) no estado de equilíbrio.
A farmacocinética do Miglustate (substância ativa) é similar em um paciente adulto com doença de Gaucher tipo 1 e em pacientes com doença de Niemann -Pick tipo C quando comparados com indivíduos saudáveis. Os dados farmacocinéticos foram obtidos em pacientes pediátricos com doença de Gaucher tipo 3 com idades de 3 a 15 anos, e pacientes com doença de Niemann Pick- tipo C com idades entre 5-16 anos.
A posologia em crianças a 200 mg três vezes ao dia ajustada à área de superfície corporal resultou em valores de Cmáx e AUC? que foram aproximadamente duas vezes superiores aos atingidos após 100 mg três vezes ao dia em pacientes com doença de Gaucher tipo 1, consistente com a farmacocinética linear a doses de Miglustate (substância ativa). No estado de equilíbrio, a concentração de Miglustate (substância ativa) no líquido cefalorraquidiano de seis pacientes com doença de Gaucher tipo 3 foi 31,4-67,2% da concentração do plasma.
Dados limitados em pacientes com doença de Fabry e com função renal comprometida mostraram que o CL/F diminui com a diminuição da função renal. Embora o número de indivíduos com insuficiência renal leve e moderada era muito pequeno, os dados sugerem uma diminuição no CL/F de aproximadamente 40% e 60 %, respectivamente, na insuficiência renal leve e moderada.
Dados em insuficiência renal grave são limitados a dois pacientes com clearance de creatinina no intervalo de 18-29 mL/min, e não podem ser extrapolados abaixo deste intervalo. Estes dados sugerem uma diminuição do CL/F de pelo menos 70% em pacientes com insuficiência renal grave. Com a gama de dados disponíveis, nenhuma relação ou tendência significativa foi observada entre os parâmetros farmacocinéticos de Miglustate (substância ativa) e variáveis demográficas (idade, IMC, sexo ou raça).
Não existe experiência com o uso de Miglustate (substância ativa) em pacientes com o doença de Gaucher tipo 1 com a idade de 18 anos. O uso de Miglustate (substância ativa) não é recomendado em crianças e adolescentes com doença de Gaucher tipo 1. Não existe experiência com o uso de Miglustate (substância ativa) em pacientes com idade superior a 70 anos.
Cuidados de Armazenamento
Zavesca deve ser armazenado em temperatura ambiente (15°C a 30°C), protegido do calor e da umidade.
O número do lote e data de fabricação são mostrados na embalagem exterior.
A data de validade é mostrada na embalagem exterior.
Este medicamento não deve ser utilizado após o prazo de validade impresso na embalagem exterior. Mantenha Zavesca em sua embalagem original.
"Antes de tomar Zavesca, verifique seus aspectos físicos."
"Todos os medicamentos devem ser mantidos fora do alcance das crianças."
Dizeres Legais
Número de Registro no Ministério da Saúde: 1.5538.0002.001-2
Farmacêutico Responsável: Fernanda Mendes – CRF/RJ 7807
Actelion Pharmaceuticals do Brasil Ltda
Rua Dalcídio Jurandir, n° 255, 3° andar Salas 306 – parte, 307 e 308, Barra da Tijuca - Rio de Janeiro – RJ, CEP 22.631-250
CNPJ 05.240.939/0001-47 – Indústria Brasileira
Atendimento ao Cliente 0800-942 0808
Fabricado por: Almac Pharma Services Limited, Armagh, Irlanda do Norte.
VENDA SOB PRESCRIÇÃO MÉDICA
Comparar preços de remédios e medicamentos no CliqueFarma é rápido e simples.
O CliqueFarma, é uma ferramenta para comparativo de preços de produtos farmacêuticos. Não comercializamos, não indicamos, não receitamos, nenhum tipo de medicamento essa função cabe exclusivamente a médicos e farmacêuticos. Não consuma qualquer tipo de medicamento sem consultar seu médico.
SE PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO. PROCURE UM MÉDICO E O FARMACÊUTICO. LEIA A BULA.
Conheça nossos Termos de Uso