- Medicamentos
- Medicamentos de A-Z
- Zemplar - Injetável 5X1Ml
Comparamos o preço de zemplar - injetável 5x1ml, veja o menor preço
Zemplar - Injetável 5X1Ml

Menor Preço
R$ 416,00
- CATEGORIA: Medicamentos de A-Z
- PRINCÍPIO ATIVO: Paricalcitol
- FABRICANTE: ABBOTT
PARA QUE SERVE?
Para que serve Zemplar é destinado para a prevenção e tratamento do hiperparatireoidismo secundário, associado à insuficiência crônica dos rins.
R
Referência
3
ofertasMelhores preços a partir de R$ 416,00 até R$ 530,55
Menor preço
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Para que serve
Zemplar é destinado para a prevenção e tratamento do hiperparatireoidismo secundário, associado à insuficiência crônica dos rins.
Como Zemplar funciona?
O hiperparatireoidismo secundário é caracterizado por um aumento do hormônio produzido pelas paratireóides – o paratormônio - associada a níveis inadequados de vitamina D. O nosso organismo obtém vitamina D através da síntese pela pele e da dieta. O paricalcitol, substância ativa do medicamento Zemplar, é uma substância semelhante à vitamina D e demonstra reduzir os níveis do hormônio paratireoidiano através da inibição da formação e secreção deste hormônio.
O tempo estimado para início da ação terapêutica deste medicamento é dependente dos níveis de hormônio paratireoidiano e da resposta individual de cada paciente. Consulte seu médico para orientação.
Contraindicação
Zemplar não deve ser administrado a pacientes com
- Evidência de toxicidade por vitamina D;
- Hipercalcemia (aumento acima do normal dos níveis de cálcio no sangue);
- Hipersensibilidade (alergia) a algum componente do produto.
Como usar
Observação: Produtos injetáveis devem ser inspecionados visualmente antes da administração, quanto a material particulado e descoloração, sempre que a solução e o recipiente permitirem. Soluções que não estejam límpidas e incolores não devem ser usadas.
Descartar as porções não utilizadas.
A via de administração usual de Zemplar é pelo mesmo acesso utilizado para hemodiálise. Para pacientes sem acesso para hemodiálise, Zemplar deve ser administrado por via intravenosa, através de injeção lenta, com pelo menos 30 segundos de duração, para minimizar a dor.
Posologia
Dose máxima
A dosafem máxima administrada com segurança é de 40 microgramas.
Dose inicial baseada na massa corpórea
A dose inicial recomendada de Zemplar é de 0,04 mcg/kg a 0,1 mcg/kg (2,8 – 7 mcg), administrada como dose in bolus, com pelo menos um dia de intervalo entre as doses, a qualquer momento durante a diálise.
Dose inicial baseada nos níveis hormonais
A dose inicial também pode ser calculada pela fórmula abaixo e administrada por via intravenosa como dose in bolus, com pelo menos um dia de intervalo entre as doses, a qualquer momento durante a diálise.
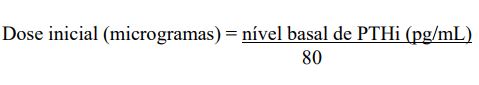
Ajuste da dose
Durante qualquer período de ajuste de dose, o cálcio e fósforo devem ser monitorados mais frequentemente e, se níveis elevados de cálcio e/ou de fósforo forem observados, a dosagem deve ser ajustada até que estes parâmetros sejam normalizados. Se hipercalcemia for observada, a dosagem de Zemplar deverá ser imediatamente reduzida ou interrompida até que este parâmetro seja normalizado. A seguir, Zemplar deve ser reiniciado com uma dose menor. Se o paciente estiver utilizando quelante de fosfato a base de cálcio, a dose deve ser diminuída ou interrompida pelo médico, ou o paciente deve trocar para um quelante de fosfato não cálcico.
As doses poderão ser reduzidas quando os níveis do hormônio da paratireóide começarem a diminuir em resposta à terapia. Assim, a dosagem deve ser individualizada.
Se uma resposta satisfatória não for observada, a dose poderá ser elevada em 2 a 4 mcg, em intervalos de duas a quatro semanas.
É importante aderir a um regime dietético de suplementação de cálcio e restrição de fósforo. Os pacientes devem ser informados sobre os sintomas da elevação de cálcio.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Zemplar?
A interrupção repentina do tratamento com esse medicamento não causa efeitos desagradáveis, apenas cessará o efeito terapêutico.
Zemplar deve ser usado sob a orientação e supervisão de um médico. A administração deste medicamento deve ser feita somente por pessoa experiente na aplicação de forma injetável de medicamentos.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
A superdosagem aguda de Zemplar pode produzir hipercalcemia (aumento acima do normal dos níveis de cálcio no sangue) e necessitar de cuidados de emergência. Durante o ajuste de dose, os níveis de cálcio e fósforo devem ser cuidadosamente monitorados pelo seu médico. Se hipercalcemia clinicamente significante se desenvolver, a dose deverá ser reduzida ou o tratamento deve ser interrompido pelo médico. A administração contínua de paricalcitol pode expor os pacientes ao risco de hipercalcemia podendo levar a calcificação vascular generalizada e outras calcificações em tecidos moles. Se ocorrer, seu médico determinará o tratamento para a diminuição do cálcio no sangue.
Fosfatos ou compostos relacionados à vitamina D não devem ser ingeridos juntamente com o paricalcitol.
A toxicidade por digitálicos é potencializada por hipercalcemia de qualquer causa; desse modo, devese ter cautela quando compostos digitálicos são prescritos juntamente a paricalcitol. Lesões ósseas adinâmicas podem se desenvolver se os níveis de do paratormônio forem suprimidos em níveis anormais.
Uso na Gravidez
Não há estudos adequados e bem controlados em mulheres grávidas. Zemplar deve ser administrado durante a gravidez apenas se os benefícios justificarem o risco potencial ao feto.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Uso na lactação
Não se sabe se o paricalcitol é excretado no leite humano. O médico lhe dará a melhor orientação quanto a decisão de descontinuar a amamentação ou descontinuar o medicamento levando-se em consideração a importância do medicamento para a mãe.
Uso Pediátrico
A segurança e a eficácia de paricalcitol em pacientes pediátricos não foram estabelecidas.
Uso em idosos
Estudos demonstraram que não houve grandes diferenças de eficácia e segurança entre pacientes com idade superior a 65 anos e pacientes mais jovens.
Insuficiência do fígado
O ajuste de dose não é necessário em pacientes com insuficiência do fígado leve a moderada. A influência da insuficiência hepática (do fígado) grave na ação de Zemplar não foi avaliada.
Insuficiência dos rins
O procedimento de hemodiálise não interfere na eliminação de Zemplar do organismo. No entanto, em comparação a voluntários sadios, pacientes com insuficiência crônica dos rins apresentam eliminação mais lenta do medicamento.
Interações medicamentosas
Estudos específicos de interação medicamentosa e seu potencial de significância não foram conduzidos com Zemplar injetável.
Deve-se ter cuidado ao administrar cetoconazol juntamente a Zemplar.
Medicamentos que contenham fosfatos ou produtos contendo vitamina D não devem ser utilizados concomitantemente a paricalcitol devido ao aumento do risco de hipercalcemia (aumento acima do normal dos níveis de cálcio no sangue).
A coadministração de altas doses de preparações contendo cálcio ou diuréticos de tiazida e paricalcitol pode aumentar o risco de hipercalcemia.
Preparações contendo magnésio (ex. antiácidos) não devem ser utilizadas em combinação com preparações de vitamina D, pois pode ocorrer hipermagnesemia (aumento acima do normal dos níveis de magnésio no sangue).
Preparações contendo alumínio (ex. antiácidos, aglutinantes de fosfato) não devem ser administrados cronicamente com produtos medicinais contendo vitamina D, pois pode ocorrer aumento dos níveis alumínio no sangue e toxicidade de alumínio nos ossos.
Durante o ajuste de dose e antes que a dose de Zemplar seja estabelecida, testes laboratoriais devem ser realizados com mais frequência.
Uma vez que a dosagem tenha sido estabelecida, cálcio e fósforo devem ser medidos no mínimo mensalmente pelo seu médico. Recomenda-se que a medição do paratormônio seja realizada a cada três meses.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
As seguintes reações adversas foram relatadas durante estudos clínicos
O evento adverso mais comum associado com a terapia de ZEMPLAR® (paricalcitol) foi a hipercalcemia (aumento acima do normal dos níveis de cálcio no sangue) que pode ser minimizado pelo ajuste de dose adequado.
Reação comum (ocorre entre 1% e 10% dos pacientes que que utilizam este medicamento)
Alterações do metabolismo e nutrição
Hipercalcemia (aumento acima do normal dos níveis de cálcio no sangue).?Alterações do sistema nervoso: disgeusia (alteração do paladar), cefaléia (dor de cabeça). Alterações gastrintestinais: hemorragia gastrintestinal (perda de sangue pelo estômago ou intestino), diarréia, constipação (prisão de ventre).
Alterações gerais e condições do local da administração
Febre, calafrios, dor no local da injeção.
Reação incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento)
Infecções e infestações
Pneumonia (infecção dos pulmões), gripe, infeção do trato respiratório superior, nasofaringite.
Neoplasias benignas e malignas (incluindo cistos e pólipos)
Câncer de mama.
Alterações do sistema linfático e hematológico
Anemia.
Alterações endócrinas
Hipoparatireoidismo (diminuição da produção do hormônio da paratireóide).
Alterações do metabolismo e nutrição
Hipocalcemia (diminuição abaixo do normal dos níveis de cálcio no sangue), hiperfosfatemia (aumento acima do normal dos níveis de fosfato no sangue), diminuição do apetite.
Alterações psiquiátricas
Delírio, estado confusional, agitação, insônia, nervosismo, inquietação.
Alterações do sistema nervoso
Acidente vascular cerebral, síncope, mioclonia (contrações repentinas, incontroláveis e involuntárias do músculo), vertigem (tonteira), hipoestesia (perda ou diminuição de sensibilidade em determinada região), parestesia (sensações cutâneas subjetivas).
Alterações visuais
Conjuntivite (inflamação dos olhos).
Alterações cardíacas
Parada cardíaca, flutter atrial (arritmias cardíacas originadas nos átrios do coração), palpitação (percepção de alteração das batidas do coração).
Alterações vasculares
Hipotensão (pressão baixa), hipertensão (pressão alta).
Alterações respiratórias, torácicas e do mediastino
Edema (inchaço) pulmonar, dispnéia (falta de ar), ortopnéia (dificuldade respiratória quando a pessoa está deitada), tosse.
Alterações gastrintestinais
Isquemia intestinal (falta de sangue no intestino), hemorragia retal, vômito, desconforto abdominal, boca seca.
Alterações de pele e tecido subcutâneo
Alopécia (perda de cabelo), rash (vermelhidão) com prurido, prurido (coceira), sensação de queimação da pele, bolhas.
Alterações musculoesqueléticas, tecido conectivo e ósseo
Artralgia (dor nas articulações), rigidez articular, mialgia (dor nos músculos), contrações musculares.
Alterações no sistema reprodutivo
Disfunção erétil (impotência), dor nas mamas.
Alterações gerais e condições do local da administração
Alterações na marcha, inchaço, astenia (fraqueza), mal estar, fadiga, condições agravadas.
Investigações
Aumento da aspartato aminotransferase, teste laboratorial anormal, perda de peso. Palpitação, hemorragia gastrointestinal, e calafrios são eventos adversos que foram observados em uma frequência maior que o placebo.
As seguintes reações adversas foram relatadas durante estudos clínicos e no período de pós- comercialização e não foi possível determinar a sua frequência
Infecções e infestações
Sépsis (infecção generalizada), infecção vaginal.
Alterações do sistema linfático e hematológico
Linfadenopatia (crescimento de um ou mais linfonodos (gânglios).
Alterações do sistema imunológico
Hipersensibilidade (alergia), angioedema (inchaço localizado e auto-limitado das camadas mais profundas da pele), edema (inchaço) de laringe.
Alterações endócrinas
Hiperparatireoidismo (aumento da produção do hormônio da paratireóide).
Alterações do metabolismo e nutrição
Hipercalemia (aumento acima do normal dos níveis de potássio no sangue).?
Alterações do sistema nervoso
Indiferença (sem resposta) ao estímulo.
Alterações visuais
Glaucoma, hiperemia ocular (aumento da quantidade de sangue nos olhos).
Alterações do ouvido e labirinto
Desconforto no ouvido.
Alterações cardíacas
Arritmia (alteração na velocidade ou ritmo do batimento cardíaco alterado).
Alterações respiratórias, torácicas e do mediastino
Chiado.
Alterações gastrointestinais
Disfagia (dificuldade de deglutição), gastrite (inflamação do estômago), náusea (enjoo).
Alterações de pele e tecido subcutâneo
Hirsutismo (crescimento excessivo de pêlos), suores noturnos, rash (vermelhidão), urticária (alergia de pele).
Alterações gerais e condições do local da administração
Desconforto no peito, dor no peito, edema (inchaço), sensação anormal, extravasamento no local da injeção, edema (inchaço) periférico, dor, sede.
Investigações
Tempo de sangramento prolongado, frequência cardíaca irregular.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária – NOTIVISA, disponível em http://www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
Composição
Cada mL de solução injetável contém:
5 mcg de paricalcitol.
Excipientes: Álcool etílico, propilenoglicol e água para injetáveis
Superdosagem
A superdosagem aguda de paricalcitol pode produzir hipercalcemia (aumento acima do normal dos níveis de cálcio no sangue), hipercalciúria (aumento acima do normal dos níveis de cálcio na urina), hiperfosfatemia (aumento acima do normal dos níveis de fosfato no sangue) e supressão elevada do hormônio da paratireóide e levar à necessidade de cuidados de emergência.
Zemplar não é significativamente removido por diálise.
Durante o ajuste de dose, os níveis de cálcio e fósforo devem ser cuidadosamente monitorados pelo médico.
Quando os níveis plasmáticos de cálcio retornam ao limite da normalidade, a terapia com Zemplar pode ser reiniciada com baixas doses. Se os níveis séricos de cálcio continuarem persistentes e acentuados, uma variedade de alternativas terapêuticas deve ser considerada pelo médico. Isto inclui o uso de drogas tais como fosfatos e corticosteroides bem como medidas para induzir a diurese (excreção de urina).
A solução de Zemplar contém 30% v/v de propilenoglicol como excipiente. Casos isolados de depressão do Sistema Nervoso Central, hemólise (destruição dos glóbulos vermelhos) e acidose lática (acúmulo de ácido láctico no corpo) foram reportados como efeitos tóxicos associados à administração de propilenoglicol em altas doses. Embora não sejam esperados com a administração de Zemplar, já que ocorre eliminação do propilenoglicol durante o processo de diálise, deve-se considerar estes efeitos em situações de superdose.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Estudos específicos de interação medicamentosa e seu potencial de significância não foram conduzidos com Paricalcitol (substância ativa) injetável.
Um estudo de interação fármaco-fármaco de múltiplas doses com cetoconazol e Paricalcitol (substância ativa) cápsulas demonstraram que cetoconazol praticamente dobrou a AUC(0-?) de Paricalcitol (substância ativa). Como o Paricalcitol (substância ativa) é parcialmente metabolizado por CYP3A e sabe-se que o cetoconazol é um forte inibidor do citocromo P450 3A, deve-se ter cautela ao administrar Paricalcitol (substância ativa) com cetoconazol ou outro forte inibidor de P450 3A.
Prescrições baseadas em fosfatos ou produtos contendo vitamina D não devem ser utilizados concomitantemente a Paricalcitol (substância ativa) devido ao aumento do risco de hipercalcemia e elevação do produto Ca X P.
A coadministração de altas doses de preparações contendo cálcio ou diuréticos de tiazida e Paricalcitol (substância ativa) podem aumentar o risco de hipercalcemia.
Preparações contendo magnésio (ex. antiácidos) não devem ser utilizadas em combinação com preparações de vitamina D pois pode ocorrer hipermagnesemia.
Preparações contendo alumínio (ex. antiácidos, aglutinantes de fosfato) não devem ser administrados cronicamente com produtos medicinais contendo vitamina D, pois pode ocorrer aumento dos níveis alumínio no sangue e toxicidade de alumínio nos ossos.
Não se espera que o Paricalcitol (substância ativa) iniba a depuração de fármacos metabolizados pelo citocromo P450, enzimas CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A; e tão pouco induza a depuração de fármacos metabolizados por CYP2B6, CYP2C9 ou CYP3A.
Ação da Substância
Resultados de eficácia
Estudos em pacientes com insuficiência renal crônica (IRC) estágio 5 mostraram que Paricalcitol (substância ativa) suprime sem diferenças significantes na incidência de hipercalcemia ou hiperfosfatemia quando comparado com o placebo. No entanto, os níveis séricos de fósforo, cálcio e o produto CaXP podem aumentar quando Paricalcitol (substância ativa) é administrado.
Em três estudos placebo-controlado1, Fase III, de 12 semanas, em pacientes com insuficiência renal crônica em diálise, Paricalcitol (substância ativa) foi introduzido a 0,04 mcg/kg, três vezes por semana. A dose foi aumentada em 0,04 mcg/kg, a cada duas semanas até que os níveis de hormônio paratireóide intacto (PTHi) diminuíssem pelo menos 30% sobre o valor basal, ou até que o quinto aumento levasse a uma dose de 0,24 mcg/kg, ou que o PTHi caísse para menos que 100 pg/mL, ou ainda, o produto Ca x P fosse maior que 75, num período de duas semanas, ou o cálcio sérico ultrapasse 11,5 mg/dL, em qualquer momento.
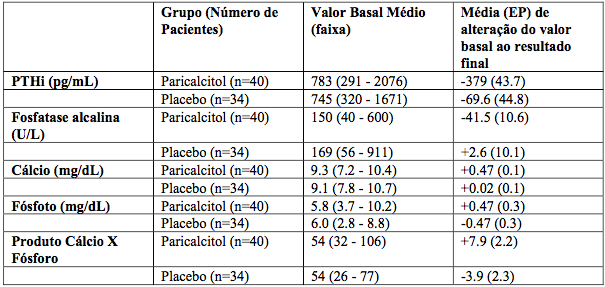
Os pacientes tratados com Paricalcitol (substância ativa) alcançaram uma redução média de PTHi de 30% em seis semanas. Nesses estudos, não houve diferença significativa na incidência de hipercalcemia ou de hiperfosfatemia entre pacientes tratados com Paricalcitol (substância ativa) e placebo. Os resultados destes estudos estão resumidos abaixo (Tabela 1):
Tabela 1: Resultados dos estudos:
Em um estudo de 12 semanas, fase IV, duplo cego, randomizado, multicêntrico, Paricalcitol (substância ativa), foi administrado em uma dose inicial de 0,04 mcg/kg ou de PTHi basal/80, três vezes por semana, para pacientes com insuficiência renal crônica (IRC estágio 5) em diálise. A dose foi aumentada em 2 mcg a cada 2 semanas até que os níveis de PTHi fossem reduzidos em 30% a 60% em relação aos níveis basais ou que o PTHi reduzisse para valores inferiores a 100pg/dL, ou o produto Ca x P aumentasse para acima de 75 por duas mensurações consecutivas, ou o cálcio sérico se elevasse para níveis superiores a 11,5 mg/dL em qualquer momento.
Os pacientes completariam o estudo se atingissem redução do PTH ? 30% em relação aos níveis basais em quatro mensurações consecutivas, ou se apresentassem um único episódio de hipercalcemia, ou completassem 12 semanas de tratamento. Não foram observados episódios de hipercalcemia em ambos os grupos de tratamento. Ambos os métodos de determinação da dose se mostraram seguros e efetivos. Os resultados estão apresentados abaixo (Tabela 2):
Tabela 2: Resultados do estudo:
| Parâmetro | PTH/80 (n=64) | 0,04 mcg/kg (n=61) |
| Incidência de hipercalcemia | 0 | 0 |
| Mediana de Dias para a Primeira de 4 Reduções do PTHi ? 30% | 31a | 45 |
| Mediana do Número de Ajustes de Doseb | 2 | 3 |
| Incidências de Ca x P > 75 | 5 (7,8%) | 2 (3,3%) |
a Estatísticamente significativo (p= 0,0306).
b Para a primeira redução de 4 ? 30% do PTHi.
Um estudo de segurança aberto de longo prazo em 164 pacientes com insuficiência renal crônica estágio 5 (dose média de 7,5 mcg três vezes por semana) demonstrou que os níveis séricos médios de Ca, P e do produto CaXP ficaram com faixas clinicamente apropriadas com a redução do PTH (redução média de 319pg/mL no 13o mês).
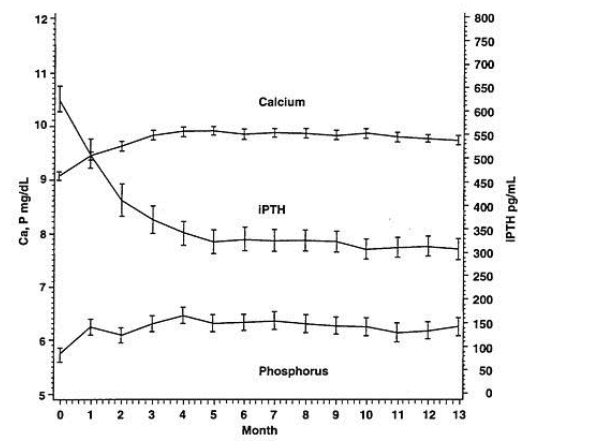
Características farmacológicas
Descrição
O Paricalcitol (substância ativa), princípio ativo de Paricalcitol (substância ativa), é um análogo sintético do calcitriol, a forma metabolicamente ativa da vitamina D com modificações na cadeia lateral (D2) e A (19-nor) do anel. O Paricalcitol (substância ativa) é um pó branco e cristalino quimicamente denominado como 19-nor-1?,3?,25- triidróxi-9,10 secoergosta-5(Z),7(E),22(E)-trieno (C27H44O3).
Propriedades Farmacodinâmicas
O hiperparatireoidismo secundário é caracterizado por uma elevação do hormônio paratireoidiano (PTH) associada a níveis inadequados de vitamina D ativa. A fonte de vitamina D no organismo é a síntese pela pele como Vitamina D3 e a dieta com Vitamina D2 e D3. Ambas as vitaminas D2 e D3 necessitam de duas hidroxilações sequenciais no fígado e nos rins para se ligar e ativar o receptor de vitamina D (VDR).
O ativador endógeno do VDR, calcitriol, é um hormônio que se liga aos VDRs presentes na glândula paratireóide, intestino, rins e ossos para manter o funcionamento da paratireóide e homeostase de cálcio e fósforo e aos VDRs que se encontram em muitos outros tecidos, incluindo próstata, endotélio e células imunes. A ativação do VDR é essencial para a formação e manutenção óssea adequadas.
Em rins deficientes, a ativação da vitamina D é diminuída, resultando no aumento de PTH e, consequentemente, levando ao hiperparatireoidismo secundário e a distúrbios na homeostase do cálcio e fósforo.
A diminuição nos níveis de calcitriol e o aumento nos níveis de PTH precedem anormalidades de cálcio e fósforo séricos e afetam a taxa de turnover ósseo, o que pode resultar em osteodistrofia renal. Em pacientes com insuficiência renal crônica, reduções no PTH estão associadas a um impacto favorável na fosfatase alcalina ósseo-específica, turnover ósseo e fibrose óssea. Além de reduzir os níveis de PTH e corrigir o turnover ósseo, a terapia com vitamina D ativa pode prevenir outras consequências da deficiência de vitamina D.
Mecanismo de ação
Estudos pré-clínicos e in vitro demonstraram que as ações biológicas do Paricalcitol (substância ativa) são medidas pela ligação com o VDR, que resulta na ativação seletiva da via de resposta da Vitamina D. Vitamina D e Paricalcitol (substância ativa) demonstraram reduzir os níveis do hormônio paratireoidiano através da inibição da síntese e secreção de PTH. Níveis reduzidos de 1,25 (OH)2D3 foram observados nos estágios iniciais da insuficiência renal crônica.
O tempo estimado para início da ação terapêutica do Paricalcitol (substância ativa) é dependente dos níveis de PTH basal e resposta individual de cada paciente.
No entanto, 3 estudos clínicos Fase III, duplo-cego, placebo-controlado, pacientes tratados com Paricalcitol (substância ativa), com dose baseada no peso corporal, atingiram uma redução média de PTHi de 30% em 6 semanas. Adicionalmente, em um estudo Fase 4, duplo-cego, duração de 12 semanas, com dose baseada tanto nos níveis de PTHi basal quanto no peso corporal, o tempo médio para a primeira entre quatro reduções ? 30% de PTHi foi de 31 dias para a dose baseada no PTHi e 45 dias para a dose baseada no peso corporal.
Farmacocinética
Duas horas após a administração de doses que variam de 0,04 a 0,24 mcg/kg, as concentrações de Paricalcitol (substância ativa) diminuíram rapidamente; depois disso, as concentrações de Paricalcitol (substância ativa) declinaram log-linearmente, com meia-vida média de cerca de 15 horas. Nenhum acúmulo de Paricalcitol (substância ativa) foi observado após doses múltiplas.
Distribuição
Paricalcitol (substância ativa) é extensamente ligado a proteínas do plasma (>99%). Em voluntários sadios, o volume de distribuição no estado de equilíbrio (steady state) é de aproximadamente 23,8 L. Após uma dose de 0,24 mcg/kg de Paricalcitol (substância ativa) em pacientes com insuficiência renal crônica estágio 5 com necessidade de hemodiálise e diálise peritoneal, a média do volume aparente de distribuição é de aproximadamente 31 a 35 L. A farmacocinética de Paricalcitol (substância ativa) foi investigada em pacientes com insuficiência renal crônica (IRC), com necessidade de hemodiálise. O Paricalcitol (substância ativa) é administrado como injeção in bolus intravenosa.
Metabolismo
Muitos metabólitos foram detectados na urina e fezes. O Paricalcitol (substância ativa) não foi detectado na urina. Dados in vitro sugerem que Paricalcitol (substância ativa) é metabolizado por várias enzimas hepáticas e não hepáticas, incluindo CYP24 mitocondrial, assim como CYP3A4 e UGT1A4.
Os metabólitos identificados incluem o produto da 24(R)-hidroxilação (presente em baixos níveis no plasma), assim como 24,26- e 24,28-diidroxilação e glicuronidação direta. Paricalcitol (substância ativa) não é um inibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A em concentrações de até 50nM (21ng/mL). Notou-se menos de duas induções com CYP2B6, CYP2C9 e CYP3A4 em concentrações semelhantes de Paricalcitol (substância ativa).
Eliminação
Paricalcitol (substância ativa) é eliminado principalmente por excreção hepato-biliar. Aproximadamente 63% da radioatividade foi eliminada nas fezes e 19% foi recuperada na urina em voluntários sadios. Nesses voluntários, a média de eliminação da meia-vida de Paricalcitol (substância ativa) é cerca de 5 a 7 horas na faixa da dose estudada de 0,04 a 0,16 mcg/kg.
Tabela 3: Parâmetros Farmacocinéticos em pacientes com insuficiência renal crônica (IRC) estágio 5 (dose única de 0,24mcg/kg in bolus intravenosa):
| IRC estágio 5-HD (n=14) | IRC estágio 5-PD (n=8)? | |
| Cmáx (ng/mL) | 1,680 ± 0,511 | 1,832 ± 0,315 |
| AUC(0-?) (ng.h/mL) | 14,51 ± 4,12 | 16,01 ± 5,98 |
| ? (1/h) | 0,050 ± 0,023 | 0,045 ± 0,026 |
| t1/2 (h)a | 13,9 ± 7,3 | 15,4 ± 10,5 |
| CL (L/h) | 1,49 ± 0,60 | 1,54 ± 0,95 |
| Vd? (L) | 30,8 ± 7,5 | 34,9 ± 9,5 |
HD: hemodiálise.
PD: diálise peritoneal.
a: média harmônica.
±: pseudo desvio padrão.
Populações especiais
Idosos
A farmacocinética de Paricalcitol (substância ativa) não foi estudada em pacientes idosos com idade superior a 65 anos.
Crianças
A farmacocinética de Paricalcitol (substância ativa) não foi estudada em pacientes com idade inferior a 18 anos.
Sexo
A farmacocinética de Paricalcitol (substância ativa) é independente do sexo.
Interações Medicamentosas
Um estudo in vitro indicou que Paricalcitol (substância ativa) não é um inibidor da CYP1A2, CYP2A6, CYP2B6, CYP 2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 OU CYP3A em concentrações acima de 50nM (21 ng/mL) (aproximadamente 20 vezes maior do que o obtido após a maior dose testada). Em culturas primárias frescas de hepatócitos, a indução observada em concentrações de Paricalcitol (substância ativa) maiores que 50nM foi menor quer duas induções para CYP2B6, CYP2C9 OU CYP3A, onde os controles positivos resultaram em indução de seis a nove vezes. Portanto, não é esperado que Paricalcitol (substância ativa) iniba ou induza a eliminação de drogas metabolizadas por estas enzimas.
Interações de injeção de Paricalcitol (substância ativa) não foram estudados.
O efeito de múltiplas doses de cetoconazol administradas como 200 mg duas vezes ao dia por cinco dias na farmacocinética de Paricalcitol (substância ativa) cápsulas foi estudada em indivíduos sadios. A Cmax de Paricalcitol (substância ativa) foi minimamente afetada, porém a AUC0-? aproximadamente dobrou na presença de cetoconazol. A meia-vida média do Paricalcitol (substância ativa) foi de 17,0 horas na presença de cetoconazol comparado com 9,8 horas quando Paricalcitol (substância ativa) é administrado isoladamente.
Cuidados de Armazenamento
Este medicamento deve ser mantido em sua embalagem original. Conservar em temperatura ambiente (15-30ºC). Proteger da luz e umidade.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características organolépticas
Zemplar apresenta-se como solução aquosa estéril, límpida e incolor.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Dizeres Legais
MS: 1.0553.0271
Farm. Resp.:
Carlos E. A. Thomazini
CRF-SP nº 24762.
Fabricado por:
Hospira SpA
Liscate - Itália
Importado por:
AbbVie Farmacêutica Ltda.
Av. Guido Caloi, 1935, 1º andar, Bloco C
São Paulo - SP
CNPJ: 15.800.545/0001-50.
Venda sob prescrição médica.
Comparar preços de remédios e medicamentos no CliqueFarma é rápido e simples.
O CliqueFarma, é uma ferramenta para comparativo de preços de produtos farmacêuticos. Não comercializamos, não indicamos, não receitamos, nenhum tipo de medicamento essa função cabe exclusivamente a médicos e farmacêuticos. Não consuma qualquer tipo de medicamento sem consultar seu médico.
SE PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO. PROCURE UM MÉDICO E O FARMACÊUTICO. LEIA A BULA.
Conheça nossos Termos de Uso