- Medicamentos
- Medicamentos Especiais
- Adcetris Liofilizado 50Mg Inj. Pó C 1 Frasco 01Brentuximab
Menor preço para Adcetris Liofilizado 50Mg Inj. Pó C 1 Frasco 01Brentuximab, você encontra no CliqueFarma
Adcetris Liofilizado 50Mg Inj. Pó C 1 Frasco 01Brentuximab

Menor Preço
R$ 19.300,00
- CATEGORIA: Medicamentos Especiais
- PRINCÍPIO ATIVO: Brentuximabe Vedotina
- FABRICANTE: TAKEDA
PARA QUE SERVE?
Para que serve Este medicamento é indicado para o tratamento de: Linfoma de Hodgkin (LH) CD30+ recidivado ou refratário que: Retornou ou nunca respondeu depois que você recebeu uma infusão de suas próprias células-tronco (transplante autólogo de células-tronco) ou retornou ou nunca respondeu a pelo menos dois tratamentos anteriores, se você não pode receber quimioterapia combinada para o câncer ou transplante autólogo de células-tronco.
R
Referência
21
ofertasMelhores preços a partir de R$ 19.300,00 até R$ 30.341,65
Menor preço
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Preço Válido para compra em Boleto
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Clique em "ir para a loja" para ir ao site da drogaria e finalize sua compra
Adcetris 50Mg, Caixa Com 1 Frasco-Ampola Com Pó Para Solução De Uso Intravenoso na Drogaria Dinâmica
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
Adcetris 50Mg 1 Frasco-Ampola Pó Para Solução De Uso Intravenoso na Sampharma Medicamentos Especiais
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Pagamento em até 6X sem juros. Frete Grátis! Regras no Site.
Para que serve
Este medicamento é indicado para o tratamento de:
Linfoma de Hodgkin (LH) CD30+ recidivado ou refratário que:
Retornou ou nunca respondeu depois que você recebeu uma infusão de suas próprias células-tronco (transplante autólogo de células-tronco) ou retornou ou nunca respondeu a pelo menos dois tratamentos anteriores, se você não pode receber quimioterapia combinada para o câncer ou transplante autólogo de células-tronco.
Linfoma anaplásico de grandes células sistêmico que é encontrado em seus linfonodos e/ou em outras partes de seu corpo que:
Nunca respondeu a outros tipos de tratamentos para o câncer ou retornou depois do último tratamento para o câncer.
O linfoma de Hodgkin e o linfoma anaplásico de grandes células sistêmico são tipos de câncer das células brancas do sangue.
Adcetris também é indicado para o tratamento de pacientes adultos com LH com risco aumentado de recidiva ou progressão após transplante autólogo de células tronco.
Como Adcetris funciona?
Adcetris contém a substância ativa brentuximabe vedotina, um agente usado no tratamento do câncer, que é composto por um anticorpo monoclonal ligado a uma substância destinada a matar as células cancerosas. Esta substância é distribuída para as células cancerosas pelo anticorpo monoclonal (uma proteína que reconhece certas células cancerosas).
Contraindicação
Este medicamento não deve ser usado por pessoas que estão usando atualmente um agente para tratar o câncer denominado bleomicina ou que apresentam hipersensibilidade (alergia) a qualquer um dos componentes do medicamento.
Como usar
Se você tiver qualquer dúvida sobre o uso deste medicamento, converse com seu médico.
A dose deste medicamento depende do seu peso corporal. A dose recomendada de Adcetris é 1,8 mg/kg, administrada uma vez a cada três semanas por no máximo 16 doses, aproximadamente por um ano, por curso de tratamento.
Caso você tenha problemas no rim ou fígado, é possível que seu médico diminua a dose inicial para 1,2 mg/kg.
Adcetris é para ser usado apenas em adultos e não deve ser usado em crianças.
Adcetris é administrado na veia (via intravenosa) na forma de infusão, durante 30 minutos, por um médico ou uma enfermeira, que irá acompanhar você durante e depois da infusão, conforme as orientações a seguir:
Instruções para a Reconstituição
Cada frasco-ampola de uso único deve ser reconstituído com 10,5 mL de água para injetáveis para obter uma concentração final de 5mg/mL.
- Direcionar o jato para a parede do frasco-ampola e não diretamente sobre a pasta ou pó.
- Girar suavemente o frasco-ampola para auxiliar a dissolução. Não agitar.
- A solução reconstituída no frasco-ampola é uma solução incolor, límpida a levemente opalescente, com pH final de 6,6.
- A solução reconstituída deve ser inspecionada visualmente para material particulado estranho e/ou descoloração. Se houver qualquer alteração, a solução deve ser descartada.
Preparação da Solução para Infusão
A quantidade apropriada de Adcetris reconstituído deve ser retirada dos frascos e adicionada a uma bolsa de infusão contendo solução injetável de cloreto de sódio 9 mg/ml (0,9%), a fim de obter uma concentração final de 0,4-1,8 mg/mL de Adcetris. O volume recomendado de diluente é 150 mL. O Adcetris já reconstituído também pode ser diluído em glicose 5% ou solução de Ringer Lactato.
Gentilmente, inverter a bolsa para misturar a solução contendo Adcetris. Não agitar.
Qualquer porção restante no frasco após a retirada do volume a ser diluído deve ser descartada de acordo com as exigências locais.
Não adicionar outros medicamentos à solução de Adcetris preparada para a infusão ou no mesmo equipo para infusão intravenosa. Após a administração, a linha de infusão deve ser lavada com solução de cloreto de sódio 0,9%, glicose 5% ou Ringer Lactato.
Após a diluição, aplicar a solução de Adcetris por infusão imediatamente, na velocidade de infusão recomendada.
O tempo total de armazenamento da solução, da reconstituição até a infusão, não deve exceder 24 horas.
Descarte
Adcetris é para uso único. Qualquer porção não usada ou sobra de material dever ser descartada de acordo com as recomendações locais.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Adcetris?
Se você não puder comparecer ao hospital no dia programado para receber o tratamento, converse com seu médico.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Ao receber o primeiro tratamento com este medicamento ou durante o curso do tratamento informe ao médico se você:
- Apresentar confusão, problemas de raciocínio, perda de memória, perda de visão ou visão borrada, força reduzida, diminuição do controle ou da sensação em um braço ou perna, alteração na maneira de andar ou perda do equilíbrio, pois estes podem ser sintomas de uma condição grave e potencialmente fatal do cérebro conhecida como leucoencefalopatia multifocal progressiva (LEMP). Se você apresentar estes sintomas antes do tratamento com este medicamento informe seu médico imediatamente sobre quaisquer alterações destes sintomas. Você também deve informar seu companheiro(a) ou cuidador sobre o seu tratamento, pois eles podem notar sintomas de que você não está ciente.
- Apresentar dor severa e persistente no estômago, com ou sem náusea e vômito. Estes sintomas podem ser decorrentes de uma condição séria e potencialmente fatal, conhecida como pancreatite (inflamação do pâncreas).
- Apresentar novos ou piora de sintomas pulmonares, como tosse ou encurtamento da respiração, uma vez que estes podem ser sintomas de uma complicação nos pulmões grave e potencialmente fatal (toxicidade pulmonar).
- Estiver tomando ou tomou anteriormente medicamentos que podem afetar o seu sistema imunológico, tais como quimioterapia ou agentes imunossupressores.
- Tem ou suspeita que tem uma infecção. Algumas infecções podem ser graves e podem ser causadas por vírus, bactérias ou outras causas que podem colocar a vida em risco.
- Sentir um som sibilante durante a respiração (respiração ofegante) ou dificuldade para respirar, ou urticária, coceira ou inchaço (sinais de uma reação à infusão).
- Tiver qualquer problema com alteração na sensibilidade da pele, especialmente nas mãos ou pés, tais como dormência, formigamento, sensação de queimação, dor, desconforto ou fraqueza (neuropatia).
- Tiver dor de cabeça, sentir cansaço, tontura, apresentar palidez (anemia) ou tiver sangramento incomum ou hematomas sob a pele, sangramento de duração maior que o normal depois que o seu sangue é coletado ou sangramento nas gengivas (redução do número de plaquetas).
- Desenvolver calafrios ou tremores ou sentir-se quente. Você deve medir a sua temperatura, pois você pode estar com febre. A ocorrência de febre com contagem baixa de glóbulos brancos do sangue pode ser um sinal de infecção grave.
- Apresentar tontura, diminuição da micção, confusão, vômito, náusea, inchaço, encurtamento da respiração ou alterações do ritmo do coração (esta pode ser uma complicação com potencial risco de vida conhecida como síndrome de lise tumoral).
- Apresentar sintomas semelhantes aos da gripe seguidos por erupção de pele dolorosa, avermelhada ou arroxeada que se espalha, e bolhas incluindo descamamento extensivo da pele que pode causar a morte (esta pode ser uma reação de pele grave conhecida como Síndrome de Stevens-Johnson e necrólise epidérmica tóxica).
- Apresentar novos ou piora de dor de estômago, náuseas, vômitos, constipação, pois estes podem ser sintomas de uma complicação estômago ou intestino grave e potencialmente fatal (complicações gastrointestinais).
- Apresentar resultados anormais de testes do fígado, pois isto pode estar relacionado a uma lesão do fígado grave e potencialmente fatal (hepatotoxicidade). Doenças do fígado e outras condições médicas que possam ter existido antes de você usar Adcetris e alguns medicamentos que você utiliza atualmente podem aumentar o risco de lesão hepática.
- Sentir-se cansado, urinar com frequência, sentir mais sede, aumento do apetite com perda de peso não intencional ou irritabilidade (hiperglicemia).
- Tiver problemas nos rins ou fígado.
Seu médico irá solicitar exames de sangue regulares para ter certeza que é seguro para você receber este medicamento.
Outros medicamentos e Adcetris:
Informe ao seu médico se você estiver usando qualquer outro medicamento, se tomou algum recentemente ou se você começar a tomar outros medicamentos, incluindo medicamentos fitoterápicos e outros medicamentos que podem ser comprados sem receita médica.
Uso durante a gravidez e a amamentação:
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico no caso de suspeita de gravidez.
Você e seu companheiro(a) devem usar dois métodos eficazes para evitar a gravidez durante o tratamento com este medicamento. As mulheres devem continuar a usar anticoncepcionais por 6 meses depois da última dose de Adcetris.
Você não deve usar Adcetris se estiver grávida exceto se você e o seu médico decidirem que o benefício para você supera o potencial risco para o seu bebê.
É importante informar ao médico antes e durante o tratamento se você estiver grávida, suspeitar que está grávida ou estiver planejando ficar grávida.
Se você estiver amamentado discuta com seu médico se você deve receber este medicamento.
Os homens em tratamento com Adcetris são aconselhados a congelar e estocar uma amostra do esperma antes de iniciar o tratamento. Aconselha-se também a não ter filhos durante o tratamento com este medicamento e por até 6 meses depois da última dose do medicamento.
Efeito na capacidade de dirigir veículos e operar máquinas:
O tratamento pode influenciar a sua capacidade de dirigir veículos ou operar máquinas. Se você não se sentir bem com o tratamento não dirija ou opere máquinas.
Informe ao seu médico ou cirurgião dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como todos os medicamentos, Adcetris pode causar efeitos colaterais, embora isso não ocorra em todas as pessoas.
Reações à infusão:
Os medicamentos deste tipo (anticorpos monoclonais) podem causar reações à infusão tais como: erupção de pele, encurtamento da respiração, dificuldade para respirar, tosse, aperto no peito, febre, dor nas costas, calafrios, dor de cabeça, vômito, náusea. As reações à infusão deste medicamento afetam mais de 1 em 10 pessoas. Em geral estes tipos de reações ocorrem dentro de minutos a várias horas depois do término da infusão. No entanto, elas podem se desenvolver mais de várias horas após o término da infusão, mas isto não é comum.
Estas reações à infusão podem ser graves ou mesmo fatais (conhecidas como reação anafilática). Não é conhecida em que frequência as reações à infusão são graves ou fatais. Você pode receber outros medicamentos como anti-histamínicos, corticosteroides ou paracetamol para auxiliar a reduzir estas reações mencionadas acima se você já tiver experimentado reações ao receber este medicamento. Se você suspeitar que já teve uma reação semelhante anteriormente, informe ao seu médico antes de receber este medicamento.
Se você desenvolver reações à infusão, seu médico pode interromper a administração deste medicamento e iniciar o tratamento de suporte. Se a infusão for reiniciada, seu médico pode aumentar o tempo durante o qual a infusão é administrada de forma a melhorar a sua tolerância ao medicamento.
Informe ao médico imediatamente se você notar algum dos sintomas descritos a seguir pois alguns deles podem possivelmente ser sinais de uma condição grave ou fatal:
Sintomas de leucoencefalopatia multifocal progressiva (LEMP) como confusão, problemas de raciocínio, perda de memória, perda de visão ou visão borrada, força reduzida, diminuição do controle ou da sensação em um braço ou perna, alteração na maneira de andar ou perda do equilíbrio. A frequência desta condição não pode ser estimada a partir dos dados disponíveis.
- Falta de ar ou tosse (afeta mais de 1 em 10 pessoas).
- Tiver qualquer alteração na sensação ou na sensibilidade, especialmente da pele, dormência, formigamento, sensação de queimação, dor, desconforto, fraqueza ou dor nas mãos ou pés (neuropatia; afeta mais de 1 em 10 pessoas).
- Sensação de fraqueza (afeta mais de 1 em 10 pessoas).
- Constipação (afeta mais de 1 em 10 pessoas).
- Diarreia, vômito (afeta mais de 1 em 10 pessoas).
- Calafrios ou tremores (afeta mais de 1 em 10 pessoas).
- Sentir-se cansado, urinar com frequência, apresentar sede aumentada, apetite aumentado com perda de peso não intencional e irritabilidade (estes podem ser sinais de hiperglicemia que afeta menos de 1 em 10 pessoas).
- Sangramento incomum ou hematomas sob a pele, sangramento de duração maior que o normal depois que o seu sangue é coletado, ou sangramento das gengivas (estes podem ser sinais de número reduzido de plaquetas no sangue que afeta menos de 1 em 10 pessoas).
- Dores de cabeça, tontura, palidez (estes podem ser sinais de anemia, que afeta menos de 1 em 10 pessoas).
- Sintomas de inflamação do pâncreas (pancreatite) como dor severa e persistente no estômago, com ou sem náusea e vômito (afeta menos de 1 em 100 pessoas).
- Apresentar sintomas semelhantes aos da gripe seguidos por erupção de pele dolorosa, avermelhada ou arroxeada que se espalha, e bolhas incluindo descamamento extensivo da pele (afeta menos de 1 em 1000 pessoas).
Você pode experimentar os seguintes efeitos colaterais:
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento):
Infecção do trato respiratório superior, neuropatia motora periférica, constipação, número diminuído dos glóbulos brancos do sangue, infecção, náusea, vômito, diarreia, coceira, ocorrência fora do normal de perda de cabelo ou pêlos, dor muscular, neuropatia sensorial periférica, cansaço, febre e reações relacionadas à infusão, dor abdominal, dor na articulação ou articulações inchadas e doloridas, calafrios, diminuição de peso, tosse e falta de ar.
Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento):
Infecção no sangue (sepse) e/ou choque séptico (uma forma de sepse que pode levar a morte), pneumonia; herpes zoster; redução do número de plaquetas no sangue; anemia; tontura; polineuropatia desmielinizante; dor nas costas; aumento do nível de açúcar no sangue; aumento no nível de enzimas do fígado e erupção cutânea (bolhas que podem ter casca ou crosta).
Reação incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento):
Síndrome de lise tumoral (uma condição com potencial risco de vida na qual você pode experimentar tontura, diminuição da micção, confusão, vômito, náusea, inchaço, encurtamento da respiração ou distúrbios do ritmo do coração); placas dolorosas, de aspecto cremoso na boca (sapinho), pneumonia por Pneumocystis jiroveci, bacteremia estafilocócica e pancreatite [sintomas de inflamação do pâncreas (pancreatite) como dor severa e persistente no estômago, com ou sem náusea e vômito].
Reação rara (ocorre entre 0,01% e 0,1% dos pacientes que utilizam este medicamento):
Síndrome de Stevens Johnson e necrólise epidérmica tóxica (um distúrbio grave e raro no qual você pode experimentar sintomas semelhantes aos da gripe seguido por uma erupção de pele dolorosa, avermelhada ou arroxeada que se espalha, e bolhas incluindo descamamento extensivo da pele).
Frequência desconhecida:
Leucoencefalopatia multifocal progressiva, neutropenia febril e reação anafilática (reação alérgica).
Atenção: Este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer reações adversas imprevisíveis ou desconhecidas. Nesse caso, informe seu médico ou cirurgião-dentista.
Composição
Cada frasco contém:
50 mg de brentuximabe vedotina.
Excipientes: Trealose di-hidratada, citrato de sódio di-hidratado, ácido cítrico monoidratado e polissorbato 80.
Superdosagem
No caso de superdose, seu médico deve monitorá-lo para controle do aparecimento de efeitos colaterais particularmente neutropenia, e tratamento de suporte será administrado, conforme a necessidade.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível.
Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Inibidores, Indutores e Substratos da CYP3A4
A coadministração Brentuximabe Vedotina (substância ativa) com cetoconazol, um inibidor potente da CYP3A4 e um inibidor da gpP, não alterou a exposição ao este medicamento; no entanto, um aumento moderado da exposição a MMAE foi observado.
Os pacientes que estão recebendo inibidores potentes da CYP3A4 e inibidores da gpP concomitantemente com este medicamento devem ser monitorados de perto para eventos adversos.
A coadministração Brentuximabe Vedotina (substância ativa) com rifampicina, um indutor potente da CYP3A4, não alterou a exposição ao este medicamento; no entanto, uma redução moderada da exposição à MMAE foi observada. Não é esperado que a coadministração deste medicamento com indutores da CYP3A4 tenha impacto na segurança ou eficácia.
A coadministração de midazolam, um substrato da CYP3A4, com este medicamento não alterou o metabolismo do midazolam; portanto, não é esperado que Brentuximabe Vedotina (substância ativa) altere a exposição aos fármacos que são metabolizados pelas enzimas CYP3A4.
Ação da Substância
Resultados de Eficácia
Linfoma de Hodgkin
Estudo: SG035-003
A eficácia e a segurança do brentuximabe vedotina como agente único foram avaliadas em um estudo, aberto, de braço único, multicêntrico em 102 pacientes com LH recidivado ou refratário.
Todos os pacientes tinham doença expressando CD30 confirmada histologicamente e tinham pelo menos um transplante autólogo de células-tronco (TACT) anterior. Setenta e dois pacientes (71%) tinham LH primário refratário, definido como uma falha em atingir uma resposta completa à terapia de primeira linha ou que progrediu dentro de 3 meses depois de completar este tratamento; 43 pacientes (42%) eram refratários e 59 pacientes (58%) tiveram recidiva depois de sua terapia prévia mais recente. Os pacientes haviam recebido uma mediana de 3,5 quimioterapias sistêmicas anteriores.
A mediana do tempo do TACT para a primeira recidiva pós-transplante foi de 6,7 meses.
Os pacientes receberam até 16 ciclos de tratamento; a mediana do número de ciclos recebidos foi igual a 9 (variando de 1 a 16). O desfecho primário, Taxa de Resposta Objetiva, foi de 74,5%. Vide a Tabela 1 abaixo para outros desfechos pré-especificados.
Tabela 1: Resultados de eficácia em pacientes com linfoma de Hodgkin reincidente ou refratário tratados com 1,8 mg/kg deste medicamento a cada 3 sem
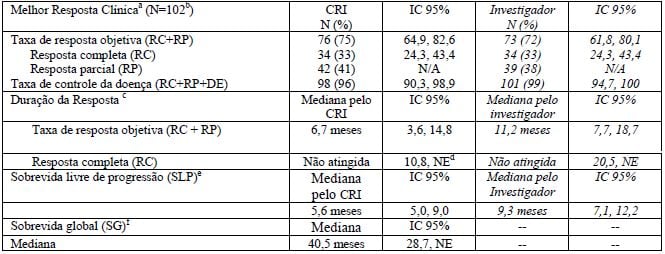
a Comitê de revisão independente (CRI) e avaliação do investigador pelos Critérios Revisados de Resposta para Linfoma Maligno (Cheson, B., Pfistner,B., Juweid, M., Gascoyne, R. & Specht, I., Horning, S., Diehl, V (2007). Revised response criteria for malignant lymphoma. Journal of Clinical Oncology, 25, 579-586. doi: 10.1200/JCO.2006.09.2403). A resposta ao tratamento foi avaliada por TC helicoidal do tórax, pescoço, abdômen e pelve; PET e dados clínicos. As avaliações da resposta foram realizadas nos ciclos 2, 4, 7, 10, 13 e 16 com PET nos ciclos 4 e 7.
b A idade dos pacientes variou de 15 a 77 anos (mediana geral, 31 anos), 53% eram do sexo feminino e 87% eram brancos; 34% dos pacientes tinham sintomas B no momento basal.
c A duração da resposta é calculada da data de resposta até a data da progressão. A mediana do tempo de seguimento desde a primeira dose foi de 9,0 meses para os pacientes que alcançaram uma resposta objetiva de acordo com o CRI.
d Não estimável.
e A mediana do tempo de seguimento (tempo para progressão da doença que ocorreu mais cedo, óbito ou último contato) desde a primeira dose foi de 5,8 meses.
f A mediana do tempo de observação (tempo para óbito ou último contato) desde a primeira dose foi de 32,7 meses.
Não foram observadas diferenças clinicamente significativas na taxa de resposta global para os subgrupos analisados, entre os seguintes subgrupos: gênero, peso inicial (? 100 kg versus > 100 kg), sintomas B no momento inicial, número de tratamentos anteriores ao TACT (? 2 versus > 2), número de tratamentos pós-TACT (0 versus ? 1), recidivado versus refratário para o último tratamento, doença refratária primária e tempo do TACT para a recidiva pós-TACT (? 1 ano versus > 1 ano). A redução do tumor ocorreu em 94% dos pacientes.
De acordo com o CRI, a mediana do tempo para a primeira resposta foi de 1,3 meses e a mediana do tempo para RC foi de 2,8 meses. A mediana da duração da resposta global foi de 6,7 meses (IC 95% [3,6; 14,8]) com variação de 1,2+ a 26,1+ meses. Dos pacientes tratados, 7 pacientes que responderam foram submetidos a um transplante alogênico de células-tronco.
Vinte e sete (77%) dos 35 pacientes que tinham sintomas B no momento inicial experimentaram resolução de todos os sintomas B em uma mediana de tempo de 0,7 meses a partir do início de Brentuximabe Vedotina (substância ativa).
Estudo SNG35-005
A eficácia e segurança do brentuximabe vedotina foram avaliadas em um estudo multicêntrico, de dois braços, controlado por placebo, duplo-cego, randomizado, em 329 pacientes com LH com risco aumentado de recidiva ou progressão após TACT. Dos 329 pacientes, 165 pacientes foram randomizados para o braço de tratamento e 164 pacientes foram randomizados para o braço de placebo. A população de segurança no braço de Brentuximabe Vedotina (substância ativa) (N = 167) incluiu dois pacientes adicionais que receberam pelo menos uma dose de ADCETRIS® mas que não foram randomizados para o braço de tratamento.
No estudo, os pacientes deveriam receber a primeira dose após a recuperação do TACT (entre os dias 30-45 após TACT). Os pacientes foram tratados com 1,8 mg/kg de Brentuximabe Vedotina (substância ativa) ou placebo correspondente por via intravenosa, durante 30 minutos a cada 3 semanas por até 16 ciclos. O número médio de ciclos recebidos em ambos os braços foi de 15 ciclos.
Os pacientes elegíveis precisavam ter, pelo menos, um dos seguintes fatores de risco:
- LH que foi refratário ao tratamento de primeira linha;
- Recidiva ou progressão de LH que ocorreu em menos de 12 meses após o fim do tratamento de primeira linha;
- Envolvimento extranodal no momento da recidiva pré-TACT, incluindo extensão extranodal de massas nodais em órgãos vitais adjacentes.
Tabela 2: Resultados de eficácia em pacientes com LH com risco aumentado de recidiva ou progressão após TACT tratados com 1,8 mg/kg de Brentuximabe Vedotina (substância ativa) a cada 3 semanas
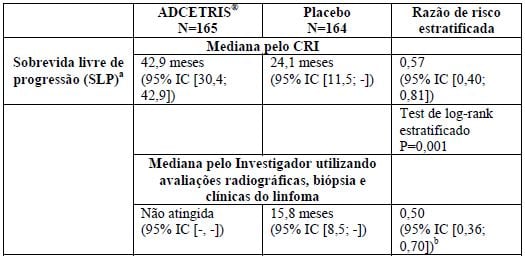
a No momento da análise, o tempo médio de acompanhamento de ambos os braços foi de 30 meses [variação, 0 a 50].
bTeste de log-rank estratificado não foi realizada para SLP por Investigador.
Análises de subgrupo pré-especificado da SLP por CRI foram realizadas por melhor resposta dos pacientes a terapia de resgate pré-TACT, estado do LH após a terapia de primeira linha, idade, sexo, peso inicial, estado do desempenho de ECOG inicial, número de tratamentos pré-TACT, região geográfica, estado do PET pré-TACT, estado do sintoma B após falha da terapia de primeira linha e estado da doença extranodal pré-TACT. As análises mostraram uma tendência consistente para benefício para os pacientes que receberam brentuximabe vedotina em comparação com pacientes que receberam placebo, com exceção dos pacientes ? 65 anos de idade (N = 8).
No momento da análise primária da SLP, uma análise interina da SG foi realizada e não houve diferença significativa na SG entre os braços tratamento e placebo. Cinquenta e três pacientes tinham morrido; 28/165 pacientes no braço brentuximabe vedotina contra 25/164 pacientes no braço do placebo.
Figura 1: Sobrevida livre de progressão por CRI (Brentuximabe Vedotina (substância ativa) vs. Placebo)
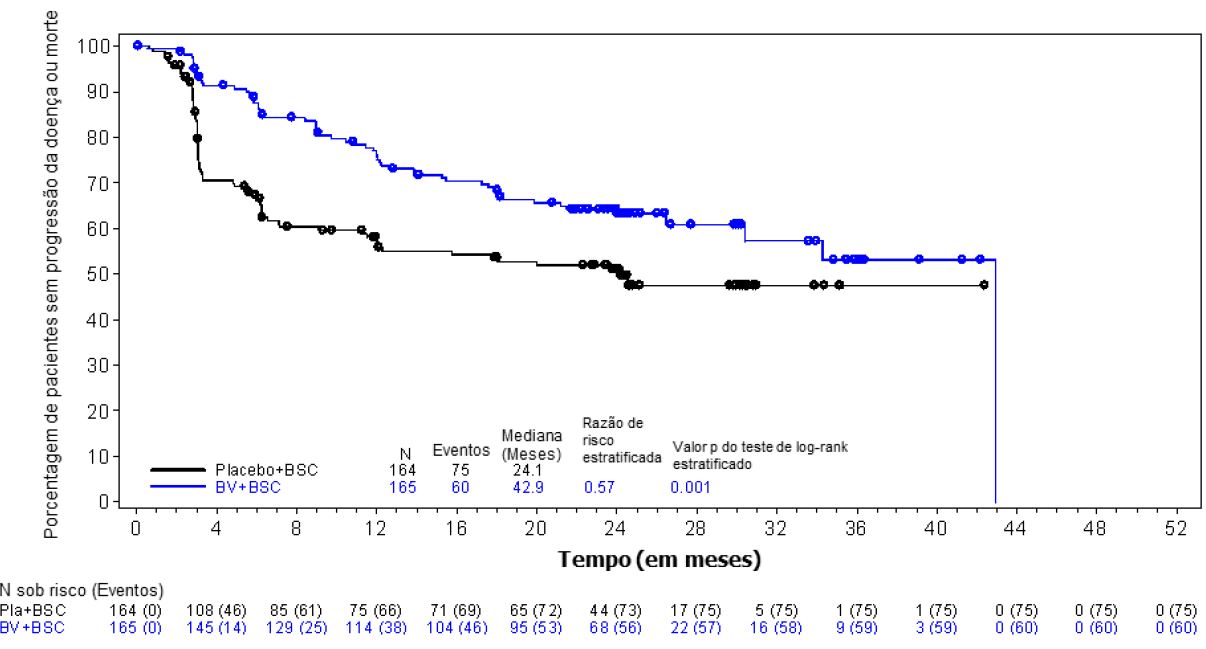
Os símbolos no gráfico indicam pacientes censurados. BV significa brentuximabe vedotina. BSC significa melhor padrão de tratamento.
Figura 2: Sobrevida livre de progressão pelo investigador utilizando avaliações radiográficas, biópsia e clínicas de linfoma (Brentuximabe Vedotina (substância ativa) vs. Placebo)
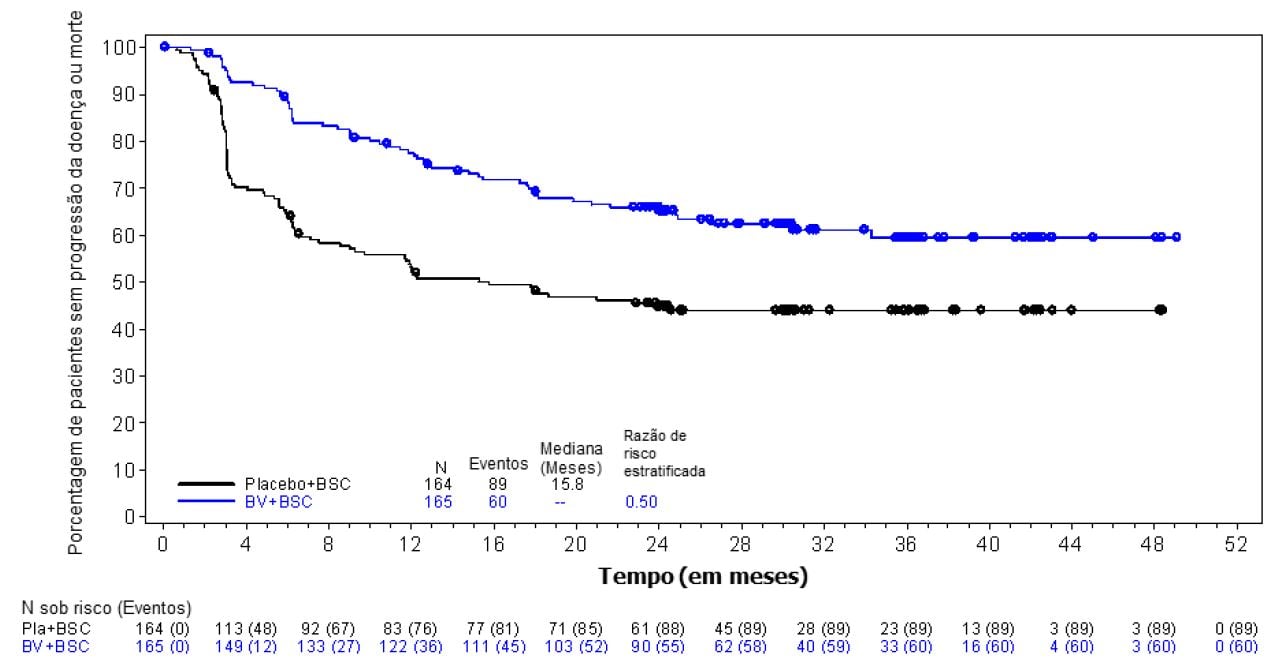
Os símbolos no gráfico indicam pacientes censurados. BV significa brentuximabe vedotina. BSC significa melhor padrão de tratamento.
Análise Post-Hoc dos Fatores de Risco
Foram realizadas análises post-hoc para avaliar o impacto do risco aumentado (número de fatores de risco) no benefício clínico (Tabela 3).
Os fatores de risco representativos para essas análises foram:
- LH que ocorreu <12 meses ou LH que foi refratário à terapia de primeira linha;
- Melhor resposta de RP ou DE para a terapia de resgate mais recente, conforme determinado pelo TC (tomografia computadorizada) e / ou PET scan;
- Doença extranodal na recidiva pré-TACT;
- Sintomas B na recidiva pré-TACT;
- Duas ou mais terapias de resgate anteriores.
Os resultados da análise post hoc sugerem aumento do benefício clínico para pacientes com dois ou mais fatores de risco, não havendo porém diferença com base em qualquer um dos fatores de risco individuais.
Nenhum benefício em termos de SLP ou SG foi observado em pacientes com um fator de risco para a recaída ou progressão.
Tabela 3: Resumo da SLP por CRI e SG por número de fatores de risco no estudo Fase 3 de LH pós-TACT
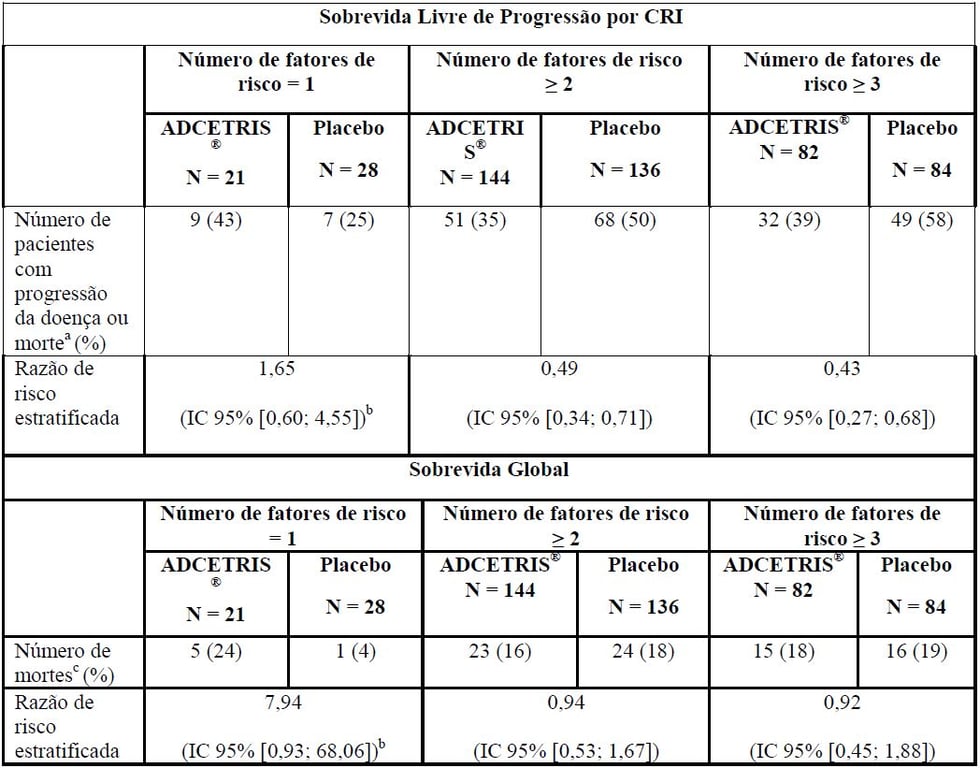
a Morte sem qualquer progressão prévia ou mais do que uma visita de avaliação perdida.
b Indica os resultados da análise não-estratificada.
c Os eventos são morte por qualquer causa.
No momento da análise atualizada (3 anos de acompanhamento) para pacientes com 2 ou mais fatores de risco, a razão de risco para SLP por CRI foi de 0,49 (IC 95% [0,34; 0,71]) e a razão de risco para SLP por investigador foi de 0,41 (IC 95% [0,29; 0,58]) (ver Figuras 1 e 2).
Estudo SNG35-006 (Estudo de Retratamento)
A eficácia do retratamento em pacientes que já haviam respondido (RC ou RP) previamente ao tratamento com brentuximabe vedotina foi avaliada em um estudo fase 2, aberto, multicêntrico. A dose inicial recomendada para o retratamento de pacientes com LH recidivado ou refratário que já haviam respondido previamente ao tratamento com brentuximabe vedotina foi 1,8 mg/kg administrada por infusão intravenosa por 30 minutos a cada 3 semanas.
Alternativamente, o tratamento pode ser iniciado com a última dose tolerada. Vinte pacientes receberam dose inicial de 1,8 mg/kg e um paciente recebeu uma dose inicial de 1,2 mg/kg de brentuximabe vedotina. O número mediano de ciclos foi 7 (variação de 2 a 37 ciclos). Dos 20 pacientes com LH avaliados, 6 pacientes (30%) alcançaram uma RC e 6 pacientes (30%) alcançaram uma RP com o retratamento com brentuximabe vedotina, para uma taxa de resposta objetiva (TRO) de 60%. A duração mediana da resposta foi 9,2 e 9,4 meses em pacientes que alcançaram RO – resposta objetiva – (RC + RP) e RC respectivamente.
Linfoma Anaplásico de Grandes Células Sistêmico
Estudo SG035-0004
A eficácia e a segurança do brentuximabe vedotina como agente único foram avaliadas em um estudo multicêntrico, de braço único, aberto, em 58 pacientes com LAGCs recidivado ou refratário.
Todos os pacientes tinham doença expressando CD30 confirmada histologicamente e haviam recebido quimioterapia de primeira linha com intenção curativa. Um total de 58 pacientes foi tratado: 36 pacientes (62%) tinham LAGCs refratário primário, definido como uma falha para alcançar uma resposta completa para a terapia de primeira linha ou progressão dentro de 3 meses após completar este tratamento; 29 pacientes (50%) eram recidivados e 29 pacientes (50%) eram refratários para a terapia anterior mais recente; 42 pacientes (72%) tinham doença negativa para quinase do linfoma anaplásico (ALK). Os pacientes haviam recebido uma mediana de 2 quimioterapias sistêmicas prévias.
Quinze pacientes (26%) haviam recebido um TACT anterior. A mediana do tempo do diagnóstico inicial de LAGCs até a primeira dose de brentuximabe vedotina foi de 16,8 meses. Os pacientes receberam até 16 ciclos de terapia; a mediana do número de ciclos recebidos foi igual a 7 (variação de 1 a 16). O desfecho primário, Taxa de Resposta Global, foi de 86,2%. Vide a Tabela 4 abaixo para outros desfechos pré-especificados.
Tabela 4: Resultados de eficácia em pacientes com LAGCs recidivado ou refratário tratados com 1,8 mg/kg de brentuximabe vedotina a cada 3 semanas
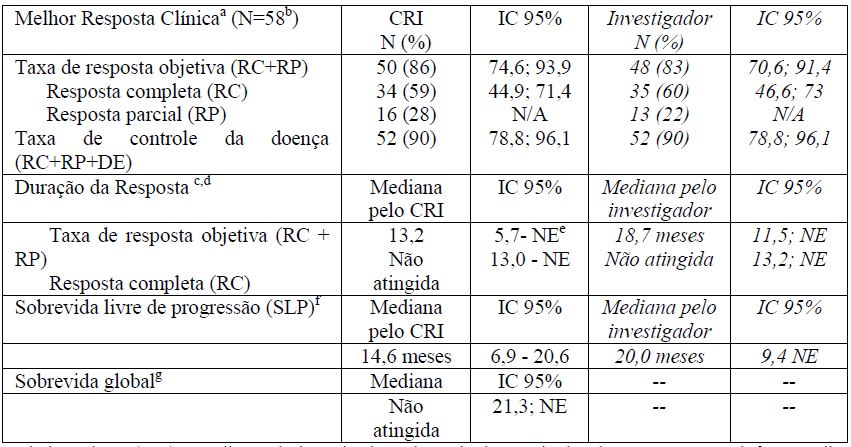
a Comitê de revisão independente (CRI) e avaliação do investigador pelos Critérios Revisados de Resposta para Linfoma Maligno (Cheson, et al. 2007). A resposta ao tratamento foi avaliada por TC helicoidal do tórax, pescoço, abdômen e pelve, PET e dados clínicos. As avaliações da resposta foram realizadas nos ciclos 2, 4, 7, 10, 13 e 16, com PET nos ciclos 4 e 7.
b A idade dos pacientes variou de 14 a 76 anos (mediana geral, 52 anos), 57% eram do sexo masculino e 83% eram brancos; 36% dos pacientes eram estadio IV ao diagnóstico inicial e 29% tinham sintomas B no momento inicial.
c A duração da resposta é calculada da data de resposta até a data da progressão. A mediana do tempo de seguimento desde a primeira dose foi de 11,8 meses para os pacientes que alcançaram uma resposta objetiva de acordo com o CRI.
d Em uma mediana de duração do tratamento de 5,4 meses e um intervalo atual de 0,7 a 17,3 meses , 27 de 50 pacientes que tiveram uma resposta objetiva haviam apresentado progressão da doença ou haviam morrido e 13 de 34 pacientes que tiveram uma RC haviam apresentado progressão da doença ou morrido.
e Não estimável
f A mediana do tempo de seguimento (tempo para progressão da doença que ocorreu mais cedo, óbito ou último contato) desde a primeira dose foi de 14,2 meses.
g A sobrevida geral estimada de 36 meses foi de 63% (IC 95% [51, 76]). A mediana de tempo de observação (tempo para óbito ou último contato) a partir da primeira dose foi de 33,4 meses.
Não foram observadas diferenças clinicamente significativas na taxa de resposta objetiva entre os seguintes subgrupos analisados: gênero, peso basal (? 100 kg versus > 100 kg), sintomas B no momento inicial, transplante autólogo de células tronco (TACT) prévio e TACT pós-tratamento. A taxa de resposta global para pacientes recidivados foi maior do que para aqueles que eram refratários (97% versus 76%).A redução do tumor ocorreu em 97% dos pacientes.
De acordo com o CRI, a mediana do tempo para a primeira resposta global foi de 1,4 meses (variação de 1,0-3,2 meses) e a mediana do tempo para resposta completa foi de 2,7 meses (variação de 1,2-11,6 meses). A mediana da duração da resposta global foi de 13,2 meses (IC 95% [5,7; NE]) com variação de 0,1+ a 21,7+ meses (a mediana do tempo de seguimento a partir da primeira dose foi de 11,8 meses). Dos pacientes tratados, 9 pacientes que responderam foram submetidos a um transplante alogênico de células-tronco e sete pacientes que responderam receberam um TACT.
Característica Farmacológica
Farmacodinâmica
Mecanismo de ação
O brentuximabe vedotina é um conjugado anticorpo-droga (CAD) composto por um anticorpo monoclonal dirigido para CD30 [imunoglobulina G1 (IgG1) quimérica recombinante produzida por tecnologia de DNA recombinante em células de ovário de hamster chinês] que está ligado covalentemente ao agente antimicrotúbulo monometil auristatina E (MMAE).
Brentuximabe Vedotina (substância ativa) libera um agente antineoplásico, seletivamente em células tumorais expressando CD30, resultando em morte celular por apoptose. Dados não clínicos sugerem que a atividade biológica de Brentuximabe Vedotina (substância ativa) resulta de um processo de etapas múltiplas. A ligação do CAD ao CD30 na superfície celular inicia a internalização do complexo CAD-CD30, que, então, se movimenta para o compartimento lisossomal. Dentro da célula, uma única parte ativa definida, MMAE, é liberada através da clivagem proteolítica. A ligação da MMAE à tubulina rompe a rede de microtúbulos dentro da célula, induz a suspensão do ciclo celular e resulta na morte, por apoptose, da célula tumoral que expressa CD30.
As contribuições para o mecanismo de ação por outras funções associadas ao anticorpo não foram excluídas.
Efeitos farmacodinâmicos
Geral
Não foram identificadas relações farmacodinâmicas primárias.
Eletrofisiologia cardíaca
Quarenta e seis (46) pacientes com doenças hematológicas malignas expressando CD30 eram avaliáveis entre os 52 pacientes que receberam 1,8 mg/kg de ADCETRIS® a cada 3 semanas como parte de um estudo de fase 1, de braço único, aberto, multicêntrico, de segurança cardíaca. O objetivo primário era avaliar o efeito do brentuximabe vedotina na repolarização ventricular cardíaca e a análise primária pré-definida foi a variação no QTc do momento basal para pontos de tempo múltiplos no Ciclo 1.
O intervalo de confiança superior a 90% (IC) foi < 10 ms em cada ponto de tempo pós-basal do Ciclo 1. Estes dados indicam a ausência de prolongamento QT clinicamente relevante devido ao brentuximabe vedotina administrado na dose de 1,8 mg/kg em pacientes com doenças malignas expressando CD30.
Farmacocinética
A farmacocinética de Brentuximabe Vedotina (substância ativa) foi avaliada em estudos de fase 1 e em uma análise da farmacocinética da população dos dados de 314 pacientes.
Absorção e biodisponibilidade
A farmacocinética sérica do CAD depois de uma dose intravenosa de Brentuximabe Vedotina (substância ativa) foi similar a de outros produtos de anticorpos. As concentrações máximas foram observadas, tipicamente, ao final da infusão ou no tempo de coleta mais próximo do final da infusão. Um declínio multiexponencial foi observado nas concentrações séricas do CAD, com meia-vida terminal de aproximadamente 4 a 6 dias. As exposições foram aproximadamente proporcionais à dose.
Depois da administração de doses múltiplas de Brentuximabe Vedotina (substância ativa), o estado de equilíbrio do CAD foi alcançado em 21 dias,consistente com a meia-vida terminal estimada. Acúmulo mínimo ou ausência de acúmulo de CAD foi observado com doses múltiplas em todos os esquemas de 3 semanas.
A eliminação da MMAE foi limitada por sua taxa de liberação do CAD. O tempo para a concentração máxima variou de aproximadamente 1 a 3 dias depois de cada infusão. As exposições da MMAE diminuíram depois de doses múltiplas de Brentuximabe Vedotina (substância ativa), com aproximadamente 50% a 80% da exposição da primeira dose sendo observados em doses subsequentes.
Distribuição
In vitro, a ligação da MMAE às proteínas do plasma humano variou de 68% a 82%. Provavelmente, a MMAE não desloca ou é deslocada por drogas altamente ligadas às proteínas. In vitro, a MMAE foi um substrato de P-gp e não foi um inibidor potente da P-gp. Em seres humanos, o volume de distribuição médio no estado de equilíbrio foi de aproximadamente 6 L a 10 L para o CAD.
Metabolismo
Os dados in vivo em animais e seres humanos sugerem que apenas uma fração pequena da MMAE liberada de Brentuximabe Vedotina (substância ativa) é metabolizada. Dados in vitro indicam que o metabolismo da MMAE que ocorre é, principalmente, via oxidação pela CYP3A4/5. Os estudos in vitro usando microssomas de fígado humano indicam que a MMAE inibe a CYP3A4/5, mas não outras isoformas. A MMAE não induziu qualquer das enzimas principais do CYP450 em culturas primárias de hepatócitos humanos.
Eliminação
Um estudo de excreção foi conduzido em pacientes que receberam uma dose de 1,8 mg/kg de Brentuximabe Vedotina (substância ativa) (brentuximabe vedotina). Aproximadamente 24% da MMAE total, administrada como parte do CAD durante uma infusão de Brentuximabe Vedotina (substância ativa), foram recuperados na urina e nas fezes durante o período de uma semana. Da MMAE recuperada, aproximadamente 72% foram recuperados nas fezes e a maior parte da MMAE excretada estava inalterada.
Uma quantidade menor da MMAE (28%) foi excretada na urina e a maior parte foi excretada inalterada.
Uso em Pediatria
Os estudos clínicos de Brentuximabe Vedotina (substância ativa) não incluíram números suficientes de pacientes com idade inferior a 18 anos para determinar se eles respondem diferentemente de indivíduos mais velhos. A segurança e a eficácia não foram estabelecidas nesta população.
Uso em Geriatria
Os estudos clínicos deBrentuximabe Vedotina (substância ativa)não incluíram números suficientes de pacientes com 65 anos de idade ou acima disso para determinar se eles respondem diferentemente dos pacientes mais jovens. A segurança e a eficácia não foram estabelecidas nesta população.
Insuficiência renal
Um estudo avaliou a farmacocinética do Brentuximabe Vedotina (substância ativa) e a MMAE após administração de 1,2 mg/kg do medicamento em pacientes com insuficiência renal leve (N=4), moderada (N=3) e grave (N=3). Comparado aos pacientes com função renal normal, a exposição de MMAE aumentou aproximadamente 1,9 vezes em pacientes com comprometimento renal grave.
Insuficiência hepática
Um estudo avaliou a farmacocinética do Brentuximabe Vedotina (substância ativa) e a MMAE após administração de 1,2 mg/kg do medicamento em pacientes com insuficiência hepática leve (Child-Pugh A; N=1), moderada (Child-Pugh B; N=5) e grave (Child- Pugh C; N=1). Comparado aos pacientes com função hepática normal, a exposição de MMAE aumentou aproximadamente 2,3 vezes em pacientes com insuficiência hepática.
Cuidados de Armazenamento
Adcetris deve ser conservado sob refrigeração (temperatura entre 2°C e 8°C), protegido da luz. Não congelar.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após a reconstituição/diluição o produto deve ser usado imediatamente ou armazenado na geladeira (temperatura entre 2°C e 8°C) e usado dentro de 24 horas.
Características físicas
Adcetris é fornecido na forma de pasta ou pó liofilizado, estéril, branco a quase branco, sem conservantes. Após a reconstituição, a solução deve ser límpida a ligeiramente opalescente, incolor e livre de partículas visíveis.
Antes de usar observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS – 1.0639.0269
Farm. Resp.:
Carla A. Inpossinato - CRF-SP nº38.535
Fabricado e embalado (emb. primária) por:
BSP Pharmaceuticals S.P.AVia Appia Km 65,561 (loc. Latina Scalo)
04013 Latina (LT) – Itália
Ou
Pierre Fabre Medicament Production
Aquitaine Pharm International 2
50 Chemin de Mazerolles
Idron, 64320 – França
Embalado (emb. secundária) por:
Takeda Italia S.p.A.
Via Crosa, 86
28065 Cerano – Itália
Ou
Takeda Austria GmbH
Linz – Áustria
Importado por:
Takeda Pharma Ltda.
Rodovia SP 340 S/N, Km 133,5
Jaguariúna - SP
CNPJ 60.397.775/0008-40
Venda sob prescrição médica.
Comparar preços de remédios e medicamentos no CliqueFarma é rápido e simples.
O CliqueFarma, é uma ferramenta para comparativo de preços de produtos farmacêuticos. Não comercializamos, não indicamos, não receitamos, nenhum tipo de medicamento essa função cabe exclusivamente a médicos e farmacêuticos. Não consuma qualquer tipo de medicamento sem consultar seu médico.
SE PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO. PROCURE UM MÉDICO E O FARMACÊUTICO. LEIA A BULA.
Conheça nossos Termos de Uso