- Medicamentos
- Medicamentos Especiais
- Imbruvica 140Mg - 90 Cápsulas Duras
Menor preço para Imbruvica 140Mg - 90 Cápsulas Duras, você encontra no CliqueFarma
Imbruvica 140Mg - 90 Cápsulas Duras

Menor Preço
R$ 20.330,00
- CATEGORIA: Medicamentos Especiais
- PRINCÍPIO ATIVO: Ibrutinibe
- FABRICANTE: JANSSEN-CILAG
PARA QUE SERVE?
Para que serve Imbruvica é um medicamento contra o câncer que contém a substância ativa ibrutinibe.
R
Referência
16
ofertasMelhores preços a partir de R$ 20.330,00 até R$ 62.772,15
Menor preço
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Preço Válido para compra em Boleto
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Para que serve
Imbruvica é um medicamento contra o câncer que contém a substância ativa ibrutinibe.
Imbruvica é usado para tratar os seguintes cânceres do sangue em adultos:
- Linfoma de Célula do Manto (LCM), um tipo de câncer que afeta os linfonodos, em pacientes que receberam no mínimo um tratamento anterior contendo rituximabe.
- Leucemia linfocítica crônica (LLC), incluindo Linfoma linfocítico de pequenas células (LLPC): câncer causado por um tipo de célula branca chamada linfócito, o qual se multiplica desordenadamente no sangue e ou nos linfonodos.
- Macroglobulinemia de Waldenström, um tipo de câncer que afeta as células brancas do sangue chamadas linfócitos, em pacientes que receberam no mínimo um tratamento para esta doença.
Como Imbruvica funciona?
Imbruvica funciona bloqueando uma proteína no corpo que ajuda as células do câncer a viver e crescer.
Esta proteína é chamada de tirosina quinase de Bruton (BKT). Através do bloqueio desta proteína, Imbruvica pode ajudar a matar e reduzir o número de células cancerosas e pode também
retardar a disseminação do câncer.
Tempo de Ação
O tempo mediano para a resposta inicial ao tratamento foi de 1,8 meses, variando de 1,4 meses a 12,2 meses em LLC/LLPC e de 1,9 meses, variando de 1,7 a 13,7 meses em LCM.
Contraindicação
Não tome Imbruvica se você for alérgico (hipersensibilidade) ao ibrutinibe ou a quaisquer dos ingredientes de sua composição.
Se você não tiver certeza disso, fale com seu médico antes de tomar Imbruvica.
Se você apresentar quaisquer sinais de reação alérgica (urticária, dificuldade para respirar, ou inchaço no seu rosto, língua ou garganta), durante o tratamento com Imbruvica, procure
socorro médico imediatamente.
Como usar
Tome Imbruvica exatamente conforme foi prescrito por seu médico ou profissional de saúde.
Não altere sua dose ou pare de tomar Imbruvica até que seu médico oriente-o a fazê-lo.
As cápsulas de Imbruvica devem ser engolidas inteiras com um copo de água. Não quebre, mastigue ou abra as cápsulas.
Tome as cápsulas de Imbruvica no mesmo horário do dia.
Imbruvica pode ser tomado antes ou após uma refeição.
Nunca dê Imbruvica a outra pessoa, mesmo que esta pessoa tenha o mesmo problema que você está sendo tratado.
Posologia
Linfoma de célula do manto (LCM)
A dose recomendada de Imbruvica é de quatro cápsulas (560 mg) via oral uma vez ao dia.
Leucemia linfocítica crônica/ Linfoma linfocítico de pequenas células ou Macroglobulinemia de Waldenström (WM)
A dose recomendada de Imbruvica é de três cápsulas (420 mg) via oral uma vez ao dia.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que devo fazer quando eu me esquecer de usar Imbruvica?
Se você esquecer de tomar uma dose, esta dose pode ser tomada assim que possível no mesmo dia, com o retorno ao esquema normal no dia seguinte. Não tome cápsulas extras para compensar a dose esquecida.
Contate seu médico se você estiver com dúvidas sobre o que fazer.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Antes de iniciar o tratamento com Imbruvica, informe seu médico ou profissional de saúde:
- Se você já teve equimose (manchas vermelho-arroxeadas na pele), ou hemorragia (sangramento) incomum ou está utilizando algum medicamento ou suplemento que aumente o risco de sangramento;
- Se você tem um histórico de batimentos cardíacos irregulares (fibrilação atrial) ou falência cardíaca severa que torna sua respiração curta e pode levar a inchaço nas pernas, ou outros problemas no coração como palpitações ou vertigens;
- Se você tem problemas no fígado ou nos rins;
- Se você fez recentemente alguma cirurgia, especialmente que possa afetar o modo de absorção de alimentos ou medicamentos no estômago ou intestino;
- Se você está planejando alguma cirurgia, seu médico pode solicitar que você interrompa o tratamento com Imbruvica por um curto período de tempo;
- Se você tem aumento de glóbulos brancos no sangue;
- Se você tem hipertensão;
- Se você está com febre ou tem alguma infecção;
- Se você tem contagem de células sanguíneas baixa;
- Se você tem ou já teve outros tipos de câncer;
- Se você apresenta grandes tumores, pois nestas situações, ao iniciar o tratamento, há o risco da ocorrência da síndrome de lise tumoral. A síndrome da lise tumoral é causada pela destruição de um grande número de células tumorais que consequentemente leva a um aumento de potássio, ácido úrico e fósforo e a uma diminuição do cálcio no sangue. Essas alterações no sangue podem afetar a função dos rins e de outros órgãos;
- Se você apresenta Síndrome congênita do QT curto ou apresenta histórico familiar de tal síndrome. Seu médico irá avaliar a prescrição de ibrutinibe para você.
Caso alguma das situações acima se aplicar a você ou você não tenha certeza, fale com seu médico ou profissional de saúde antes de tomar Imbruvica.
Exames laboratoriais antes e durante o tratamento
Exames de laboratório podem demonstrar que sua contagem sanguínea possui mais células brancas (chamadas ‘linfócitos’), nas primeiras semanas de tratamento (linfocitose). Isto é esperado e pode permanecer por alguns meses. Isto não significa necessariamente que o seu câncer do sangue piorou.
Seu médico verificará sua contagem sanguínea antes ou durante o tratamento e em casos raros pode ser necessário administrar outro medicamento.
Fale com seu médico sobre o significado dos resultados dos seus exames.
Uso pediátrico e em adolescentes
Imbruvica não deve ser usado em crianças e adolescentes abaixo de 18 anos de idade, pois não há estudos do medicamento nesta faixa etária.
Imbruvica e outros medicamentos
Fale com seu médico ou profissional de saúde se você está tomando, tomou recentemente, ou possa vir a tomar algum outro medicamento. Isto inclui medicamentos isentos de prescrição médica, fitoterápicos e suplementos.
Isto porque Imbruvica pode afetar o funcionamento de outros medicamentos. Além disso, outros medicamentos podem afetar o funcionamento de Imbruvica.
Imbruvica pode aumentar o risco de sangramento
Fale com seu médico ou profissional de saúde se você toma outros medicamentos que aumentam os riscos de sangramento, incluindo:
- Ácido acetilsalicílico (AAS, Aspirina) e anti-inflamatórios não esteroidais, como ibuprofeno ou naproxeno;
- Anticoagulantes tais como varfarina, heparina ou outras medicações para prevenir ou tratar a formação de coágulos sanguíneos;
- Suplementos que podem aumentar o risco de sangramento tais como óleo de peixe e vitamina E.
Os efeitos de Imbruvica ou outros medicamentos podem ser influenciados se você toma Imbruvica em conjunto com qualquer um dos medicamentos a seguir.
Informe seu médico se você toma:
- Antibióticos para tratar infecções bacterianas, como claritromicina, telitromicina, ciprofloxacino, eritromicina ou rifampicina;
- Medicamentos para tratar infecções fúngicas, como cetoconazol, itraconazol, fluconazol, voriconazol ou posaconazol;
- Medicamentos para tratar infecção por HIV, como ritonavir, cobicistate, indinavir, nelfinavir, saquinavir, amprenavir, atazanavir, ou fosamprenavir;
- Medicamentos para prevenir náusea e vômitos associados com quimioterapia, como aprepitanto;
- Medicamentos para tratar depressão, como nefazodona;
- Medicamentos chamados inibidores de quinase para tratamento de outros cânceres, como crizotinibe e imatinibe;
- Medicamentos chamados de bloqueadores de canal de cálcio para hipertensão (pressão alta) ou dor no peito, como diltiazem, verapamil;
- Medicamentos chamados estatinas para tratar colesterol alto como rosuvastatina;
- Medicamentos para o coração/antiarrítmicos como amiodarona, dronedarona;
- Medicamentos para prevenir convulsões ou tratar epilepsia ou medicamentos para tratar condições dolorosas na face chamadas nevralgia trigeminal, como carbamazepina e fenitoína;
- Um fitoterápico usado, por exemplo, para depressão, como erva de São João.
Se você está tomando digoxina, um medicamento para problemas do coração, ou metotrexato, um medicamento usado para tratar outros canceres e para reduzir a atividade do sistema imunológico (por exemplo, para artrite reumatoide ou psoríase), eles devem ser tomados pelo menos 6 horas antes ou depois de Imbruvica.
Contate seu médico se tiver alguma dúvida se seu medicamento está listado acima. Conheça os medicamentos que você toma. Tenha uma lista para mostrar ao seu médico ou profissional de saúde quando você receber um novo medicamento.
Imbruvica com alimento
Não tome Imbruvica com toranja (‘grapefruit’) ou Laranjas de Sevilha. Isto inclui comê-la, tomar o suco ou suplementos que possam contê-la, pois ela pode aumentar a quantidade de
Imbruvica em seu sangue.
Efeito sobre a capacidade de dirigir ou operar máquinas
Você pode sentir fadiga (cansaço) ou tontura e astenia (ausência ou diminuição da força física) tomando Imbruvica e isso pode afetar sua capacidade para dirigir, usar qualquer ferramenta e operar máquinas.
Gravidez, amamentação e fertilidade
Você não deve engravidar enquanto estiver tomando Imbruvica.
Se você está grávida, acha que está grávida ou planeja ter um bebê, converse com seu médico ou profissional de saúde para devida orientação antes de tomar Imbruvica.
Imbruvica não deverá ser utilizado durante a gestação. Não há informação sobre a segurança de Imbruvica em mulheres grávidas.
Mulheres em idade fértil devem usar métodos contraceptivos eficazes para evitar gravidez enquanto estiver tomando Imbruvica e por pelo menos um mês após o tratamento com Imbruvica.
Se você estiver utilizando métodos hormonais, como por exemplo, pílula anticoncepcional ou dispositivos para evitar a gravidez, você deve adicionar um método de barreira (por exemplo, preservativo). Não se sabe quanto tempo após o tratamento com Imbruvica é seguro engravidar.
Informe seu médico imediatamente se você ficar grávida.
Não amamente enquanto estiver tomando Imbruvica.
Os homens não devem conceber um filho ou doar esperma enquanto estiverem tomando Imbruvica e por 3 meses após a conclusão do tratamento.
Use preservativos e não doe esperma durante e por 3 meses após a conclusão do tratamento. Se você planeja ser pai, fale com seu médico ou profissional de saúde antes de tomar Imbruvica.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Informe imediatamente seu médico em caso de suspeita de gravidez.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como todos os medicamentos, Imbruvica pode causar efeitos adversos. Os seguintes efeitos adversos podem acontecer tomando este medicamento:
Hemorragia (sangramento)
Você pode apresentar equimose (manchas vermelho arroxeadas na pele) ou sangramentos nasais durante o tratamento com Imbruvica. Raramente, podem ocorrer sangramentos internos graves, como sangramento no estômago, intestino ou cérebro, algumas vezes ocasionando a morte.
Fale com seu médico ou profissional de saúde se você apresentar sinais ou sintomas de sangramento grave, como sangue nas fezes ou urina e sangramento que dure mais tempo ou que você não pode controlar.
Leucostase (dificuldade na circulação do sangue devido ao aumento do número de células brancas no sangue)
Você pode apresentar um aumento no número de células brancas no sangue, especialmente linfócitos. Em casos raros este aumento pode ser severo, causando agregação das células, dificultando o fluxo do sangue pelos vasos sanguíneos. Seu médico irá monitorar sua contagem sanguínea.
Infecções
Você pode apresentar infecções virais, bacterianas ou fúngicas durante o tratamento com Imbruvica.
Fale com seu médico se você tiver febre, calafrios, fraqueza, confusão, dores no corpo, frio ou sintomas de resfriado, sentir-se cansado ou sentir falta de ar. Estes podem ser sinais de infecção.
Redução na contagem de células do sangue
O uso de Imbruvica pode levar a um número baixo de células vermelhas do sangue (anemia), número baixo de um tipo de célula branca do sangue (neutrófilo) ou plaquetas (células que ajudam o sangue a coagular). Seu médico ou profissional de saúde deve verificar sua contagem de células regularmente.
Doença pulmonar Intersticial (DPI)
Inflamação dentro dos pulmões que pode levar a dano permanente aconteceu com o tratamento com Imbruvica. Fale com seu médico se você tiver dificuldade para respirar ou tiver tosse persistente.
Problemas no coração
Batimentos irregulares no coração (fibrilação atrial) podem acontecer durante o tratamento com Imbruvica. Informe seu médico ou profissional de saúde se você tem problemas cardíacos.
Síndrome de lise tumoral
Níveis não usuais de substâncias químicas no sangue causadas pela rápida morte de células cancerosas aconteceram durante o tratamento de câncer e às vezes mesmo sem tratamento. Isto pode levar a alterações na função dos rins, batimentos cardíacos anormais ou convulsões.
Seu médico ou outro profissional de saúde pode realizar testes de sangue para checar a síndrome de lise tumoral.
Cânceres de pele não melanoma
Tipos de cânceres de pele que não são melanoma, mais frequentemente cânceres de células escamosas ou de célula basal, aconteceram com pessoas em tratamento com Imbruvica.
Reações alérgicas
Interrompa o uso de Imbruvica e procure atendimento médico se você tiver qualquer um desses sinais de reação alérgica:
Rosto, lábios, boca, língua ou garganta inchados, dificuldade de engolir ou respirar, erupções cutâneas com coceira (urticária), vermelhidão na pele.
As reações adversas mais comuns em pessoas com Linfoma de células do manto (? 20%) incluem:
Diarreia, hemorragia [por exemplo: equimose (manchas vermelho arroxeadas na pele)] cansaço (fadiga), dor musculoesquelética, náusea, infecção do trato respiratório superior (infecção de nariz, seios da face ou garganta), tosse e erupção cutânea.
As reações adversas descritas a seguir foram coletadas em estudos clínicos com Imbruvica, para o tratamento de Linfoma de célula do manto (LCM):
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento)
- Pneumonia;
- Infecção do trato urinário;
- Infecção do trato respiratório superior;
- Sinusite (infecção dos seios da face);
- Neutropenia (diminuição de neutrófilos, um tipo de células brancas do sangue);
- Trombocitopenia (diminuição do número de plaquetas no sangue, células que ajudam o sangue a coagular);
- Anemia (diminuição de hemácias, as células vermelhas do sangue);
- Desidratação;
- Hiperuricemia (níveis de ácido úrico elevada no sangue);
- Diminuição do apetite;
- Tontura;
- Dor de cabeça;
- Dispneia (dificuldade de respirar ou respiração curta);
- Epistaxe (sangramento do nariz);
- Tosse;
- Diarreia;
- Dor abdominal;
- Vômito;
- Estomatite (aftas na boca e/ou garganta);
- Constipação (prisão de ventre);
- Náusea;
- Dispepsia (indigestão);
- Erupção cutânea;
- Espasmos musculares (contração involuntária dos músculos);
- Mialgia (dor muscular);
- Artralgia (dor articular);
- Dor nas costas;
- Dor nas extremidades;
- Pirexia (febre);
- Fadiga (cansaço);
- Astenia (fraqueza generalizada, ausência ou diminuição de força física);
- Edema periférico (mãos, tornozelos ou pés inchados);
- Conjuntivite (olhos vermelhos por inflamação da conjuntiva).
Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento)
- Pneumonia;
- Infecção do trato urinário;
- Desidratação;
- Hiperuricemia (níveis de ácido úrico elevada no sangue);
- Diminuição do apetite;
- Dispneia (dificuldade de respirar ou respiração curta);
- Diarreia;
- Dor abdominal;
- Infecção de pele;
- Fadiga (cansaço);
- Astenia (fraqueza generalizada, ausência ou diminuição de força física);
- Edema periférico (mãos, tornozelos ou pés inchados);
- Contusão (hematomas);
- Fibrilação atrial (batimentos cardíacos irregulares).
Reação incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento)
- Sinusite (inflamação dos seios da face);
- Dor nas costas;
- Náusea;
- Estomatite (infecção viral que provoca várias aftas na boca e garganta);
- Pirexia (febre).
As reações adversas graves/com risco de morte mais comuns (? 5%) foram:
Neutropenia (diminuição de neutrófilos, um tipo de células brancas do sangue), trombocitopenia (diminuição do número de plaquetas no sangue), pneumonia e anemia (diminuição de hemácias, as células vermelhas do sangue).
As reações adversas mais frequentes levando à descontinuação do tratamento incluíram hemorragia, pneumonia e trombocitopenia (diminuição do número de plaquetas no sangue).
As reações adversas graves foram reportadas em 60% dos pacientes (frequências de tratamento emergente).
Reações adversas graves que ocorreram em mais de 2% dos pacientes foram fibrilação atrial (batimentos cardíacos irregulares), pneumonia, infecção do trato urinário, dor abdominal, hematoma subdural (acúmulo de sangue entre o cérebro e o crânio), neutropenia febril (diminuição de neutrófilos no sangue com febre), insuficiência renal aguda, edema periférico (inchaço nas mãos, tornozelos ou pés) e pirexia (febre).
Reações adversas provenientes de outro estudo clínico
As reações adversas de ocorrência mais comum (? 20%,) nos estudos clínicos para o tratamento de Leucemia linfocítica crônica/linfoma linfocítico de pequenas células que receberam pelo menos uma terapia anterior foram: diarreia, dor musculoesquelética, infecção do trato respiratório superior, equimoses (manchas vermelho arroxeadas na pele), erupção cutânea, náusea, febre, neutropenia (baixo número de um tipo de células brancas do sangue) e anemia (baixo número de células vermelhas do sangue).
Se você tiver diarreia que dure mais de uma semana, seu médico poderá prescrever reposição de líquido e sal ou outro medicamento.
As reações adversas descritas a seguir foram coletadas em estudos clínicos com Imbruvica, para o tratamento de Leucemia linfocítica crônica/linfoma linfocítico de pequenas células que receberam pelo menos uma terapia anterior:
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento):
- Infecção do trato respiratório superior;
- Pneumonia;
- Sinusite;
- Anemia (diminuição de hemácias, as células vermelhas do sangue);
- Neutropenia (diminuição de neutrófilos, um tipo de células brancas do sangue);
- Trombocitopenia (diminuição do número de plaquetas no sangue);
- Dor de cabeça;
- Tontura;
- Diarreia;
- Náusea;
- Estomatite (aftas na boca e/ou garganta);
- Constipação (prisão de ventre);
- Vômito;
- Equimose (manchas vermelho-arroxeadas na pele);
- Erupção cutânea;
- Petéquias (pontos vermelhos no corpo);
- Dor musculoesquelética;
- Artralgia (dor articular);
- Pirexia (febre).
Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento)
- Infecção do trato urinário;
- Infecção de pele;
- Sepse (infecção generalizada);
- Linfocitose (aumento no número de um tipo de células brancas do sangue, os linfócitos);
- Leucocitose (aumento no número de células brancas do sangue);
- Neutropenia febril (diminuição de neutrófilos no sangue com febre);
- Visão turva;
- Fibrilação atrial (ritmo cardíaco anormal);
- Epistaxe (sangramento do nariz);
- Hematoma subdural (acúmulo de sangue entre o cérebro e o crânio).
Reações adversas graves/com risco de morte mais comuns (?5%):
Neutropenia, pneumonia, trombocitopenia (diminuição do número de plaquetas no sangue) e neutropenia febril (diminuição de neutrófilos no sangue com febre).
As reações adversas mais frequentes que causaram descontinuação do tratamento incluem infecções, diarreia, e hematoma subdural (acúmulo de sangue entre o cérebro e o crânio).
As reações adversas graves ocorreram mais frequentemente entre pacientes idosos.
Aproximadamente 9% dos pacientes com LLC/LLPC que receberam pelo menos uma terapia anterior, recebendo ImbruvicaTM nos estudos clínicos, descontinuaram o tratamento devido a reações adversas. Estas incluíram infeções, diarreia e hematomas subdurais. As reações adversas que causaram redução de dose ocorreram em aproximadamente 6% dos pacientes.
As reações adversas de ocorrência mais comum nos estudos de Macroglobulinemia de Waldenström (MW) foram: neutropenia, trombocitopenia, diarreia, erupção cutânea, náusea, espasmos musculares e fadiga.
As reações adversas descritas a seguir foram coletadas em estudos clínicos com Imbruvica, para o tratamento Macroglobulinemia de Waldenström (MW).
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento)
- Sinusite (infecção dos seios da face);
- Infecção do trato respiratório superior;
- Pneumonia;
- Infecção cutânea;
- Câncer de pele;
- Neutropenia (diminuição de neutrófilos, um tipo de células brancas do sangue);
- Trombocitopenia (diminuição do número de plaquetas no sangue, células que ajudam o sangue a coagular);
- Anemia (diminuição de hemácias, as células vermelhas do sangue);
- Tontura;
- Cefaleia (dor de cabeça);
- Epistaxe (sangramento do nariz);
- Tosse;
- Diarreia;
- Náusea;
- Estomatite (aftas na boca e/ou garganta);
- Doença do refluxo gastroesofágico (refluxo de conteúdo alimentar presente no estômago para o esôfago);
- Erupção cutânea;
- Equimoses;
- Prurido;
- Espasmos musculares (contração involuntária dos músculos);
- Artropatia (doença das articulações);
- Fadiga (cansaço).
As reações adversas graves/com risco de morte (pneumonia, fibrilação atrial e infecção do trato urinário), ocorreram mais frequentemente entre pacientes idosos tratados com Imbruvica.
Entretanto deve-se enfatizar que muitas pessoas não terão nenhum desses problemas. Não hesite em relatar qualquer efeito indesejável ao seu médico ou farmacêutico. Além disso, informe seu médico ou farmacêutico se você notar qualquer efeito adverso não mencionado nesta bula.
Dados de Pós-comercialização (frequência estimada a partir de relato espontâneo)
Insuficiência hepática (problemas no fígado), síndrome de lise tumoral, angioedema (inchaço na pele), eritema (vermelhidão na pele) e urticária foram reportados durante a experiência pós-comercialização como reações adversas muito raras. A Síndrome de Stevens-Johnson (erupção cutânea severa com bolhas e descamação da pele, particularmente ao redor da boca, nariz, olhos e genitais) foi reportada como reação adversa rara. Doença Pulmonar Intersticial e onicoclasia (ruptura das unhas) foram reportadas como reações adversas incomuns.
Atenção: Este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
Composição
Cada cápsula gelatinosa dura contém:
140 mg de ibrutinibe.
Excipientes: croscarmelose sódica, estearato de magnésio, celulose microcristalina, laurilsulfato de sódio, gelatina e dióxido de titânio.
Superdosagem
Se você tomar mais cápsulas de Imbruvica do que deveria, contate seu médico ou vá ao hospital imediatamente.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível.
Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Ibrutinibe (substância ativa) é metabolizado primariamente pelo citocromo P450 enzima 3A (CYP3A).
Agentes que podem aumentar as concentrações plasmáticas de Ibrutinibe (substância ativa)
O uso concomitante de Ibrutinibe (substância ativa) e medicamentos que inibem de modo potente ou moderado a CYP3A pode aumentar a exposição ao Ibrutinibe (substância ativa), devendo ser evitado os inibidores potentes da CYP3A.
Inibidores potentes da CYP3A
A administração concomitante de cetoconazol, um potente inibidor da CYP3A, em 18 indivíduos saudáveis, aumentou a exposição (Cmáx e AUC0-última) ao Ibrutinibe (substância ativa) em 29 e 24 vezes, respectivamente. A exposição máxima observada ao Ibrutinibe (substância ativa) (AUC) foi ? 2 vezes em 37 pacientes tratados com inibidores leves e/ou moderados da CYP3A em comparação à exposição a Ibrutinibe (substância ativa) em 76 pacientes não tratados concomitantemente com inibidores da CYP3A.
Os dados de segurança clínica em 66 pacientes tratados com inibidores moderados (n=47) ou potentes da CYP3A (n=19) não revelaram aumentos significativos nas toxicidades. Inibidores potentes da CYP3A (por exemplo, cetoconazol, indinavir, nelfinavir, ritonavir, saquinavir, claritromicina, telitromicina, itraconazol, nefazodona, cobicistate e posaconazol) deverão ser evitados. Caso o benefïcio supere o risco e um inibidor potente da CYP3A deva ser utilizado, reduzir a dose de Ibrutinibe (substância ativa) para 140 mg pelo período de uso do inibidor ou suspender temporariamente o tratamento com Ibrutinibe (substância ativa) (por 7 dias ou menos).
Inibidores moderados e leves da CYP3A
Em pacientes com neoplasias malignas de célula B, a coadministração de inibidores da CYP3A, eritromicina e voriconazol aumentou a Cmáx em 3,4 vezes e 6,7 vezes e aumentou a AUC em 3,0 vezes e 5,7 vezes, respectivamente. Caso um inibidor moderado da CYP3A (por exemplo, fluconazol, voriconazol, eritromicina, amprenavir, aprepitanto, atazanavir, ciprofloxacino, crizotinibe, diltiazem, fosamprenavir, imatinibe, verapamil, amiodarona, dronedarona) seja indicado, reduzir a dose de Ibrutinibe (substância ativa) para 140 mg pelo período de uso do inibidor.
Não é requerido ajuste de dose em combinação com inibidores leves. Monitorar o paciente rigorosamente quanto à toxicidade e seguir a orientação de modificação de dose conforme necessário. Evitar suco de toranja (‘grapefruit’) e laranjas de Sevilha durante o tratamento com Ibrutinibe (substância ativa) , uma vez que estes contêm inibidores moderados da CYP3A.
Agentes que podem reduzir as concentrações plasmáticas de Ibrutinibe (substância ativa)
A administração de Ibrutinibe (substância ativa) com indutores potentes da CYP3A reduz as concentrações plasmáticas de Ibrutinibe (substância ativa) em até 90%.
Evitar o uso concomitante de indutores potentes da CYP3A (por exemplo, carbamazepina, rifampina, fenitoína e erva de São João). Considerar agentes alternativos com menor indução de CYP3A.
Medicamentos que podem ter suas concentrações plasmáticas alteradas pelo Ibrutinibe (substância ativa)
Estudos in vitro indicaram que o Ibrutinibe (substância ativa) é um inibidor fraco reversível de CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4/5 e não revela inibição dependente de tempo para CYP450. O metabólito di-hidrodiol de Ibrutinibe (substância ativa) é um inibidor fraco de CYP2B6, CYP2C8, CYP2C9 e CYP2D6. O Ibrutinibe (substância ativa) e o metabólito di-hidrodiol são no máximo indutores fracos das isoenzimas CYP450 in vitro. Portanto, é improvável que Ibrutinibe (substância ativa) possua quaisquer interações medicamentosas clinicamente relevantes com medicamentos que podem ser metabolizados pelas enzimas CYP450.
Estudos in vitro indicaram que o Ibrutinibe (substância ativa) não é substrato da P-gp nem de outros transportadores importantes, exceto OCT2. O metabólito di-hidrodiol e outros metabólitos são substratos de P-gp. O Ibrutinibe (substância ativa) é um inibidor leve de P-gp e da proteína de resistência ao câncer de mama (PRCM). Não se espera que Ibrutinibe (substância ativa) possua interações medicamentosas sistêmicas com substratos da P-gp. Entretanto, não se pode excluir que Ibrutinibe (substância ativa) possa inibir a P-gp intestinal e a PRCM após uma dose terapêutica. Não há dados clínicos disponíveis.
Para minimizar o potencial de uma interação no trato gastrintestinal, subtratos da P-gp ou PRCM com intervalo terapêutico estreito, como a digoxina ou metotrexato, devem ser administrados pelo menos 6 horas antes ou depois de Ibrutinibe (substância ativa). O Ibrutinibe (substância ativa) também pode inibir sistematicamente a PRCM e aumentar a exposição de medicamentos que são submetidos ao efluxo hepático mediado pela PRCM, como a rosuvastatina.
Ação da Substância
Resultados de eficácia
Linfoma de célula do manto
A segurança e a eficácia de Ibrutinibe (substância ativa) em pacientes com LCM que receberam no mínimo um tratamento anterior foram avaliadas em um único estudo de Fase 2, aberto, multicêntrico (PCYC-1104- CA) de 111 pacientes. A idade mediana foi de 68 anos (variação, 40 a 84 anos), 77% eram do sexo masculino e 92% eram caucasianos. O tempo mediano desde o diagnóstico foi 42 meses, e o número mediano de tratamentos anteriores foi 3 (variação, 1 a 5 tratamentos), incluindo 35% com uso prévio de quimioterapia de altas doses, 43% com uso prévio de bortezomibe, 24% com uso prévio de lenalidomida e 11% com transplante prévio de céluas estaminais (células tronco). Na visita basal, 39% dos pacientes apresentaram doença volumosa (? 5 cm), 49% tinham pontuação de alto risco pelo Índice de Prognóstico Internacional simplificado para LCM (MIPI) e 72% tinham doença avançada (envolvimento extranodal e/ou de medula óssea) na triagem.
Ibrutinibe (substância ativa) foi administrado por via oral a 560 mg uma vez ao dia até a progressão da doença ou toxicidade inaceitável. A resposta do tumor foi avaliada de acordo com os critérios revisados do Grupo Internacional (IWG) de linfoma não Hodgkin (LNH). O desfecho primário neste estudo foi a taxa de resposta global avaliada pelo investigador (TRG). As respostas de Ibrutinibe (substância ativa) estão demostradas na tabela a seguir:
Taxa de resposta global e duração da resposta baseada na avaliação do investigador em pacientes com linfoma de célula do manto
|
Total N=111 |
|
|
Taxa de resposta global (%) |
67,6 |
|
IC de 95% |
(58,0; 76,1) |
|
RC (%) |
20,7 |
|
RP (%) |
46,8 |
|
Mediana da duração da resposta (RC+RP) (meses) |
17,5 (15,8, NA) |
|
Tempo mediano para resposta inicial, meses (variação) |
1,9 (1,4-13,7) |
|
Tempo mediano para resposta completa, meses (variação) |
5,5 (1,7-11,5) |
IC = intervalo de confiança.
RC = resposta completa.
RP = resposta parcial.
NA: não atingido.
Os dados de eficácia foram adicionalmente avaliados por um Comitê de Revisão Independente demostrando uma taxa de resposta global de 69% com 21% de taxa de resposta completa e 48% de taxa de resposta parcial. A mediana da duração da resposta estimada pelo Comitê de Revisão Independente foi 19,6 meses.
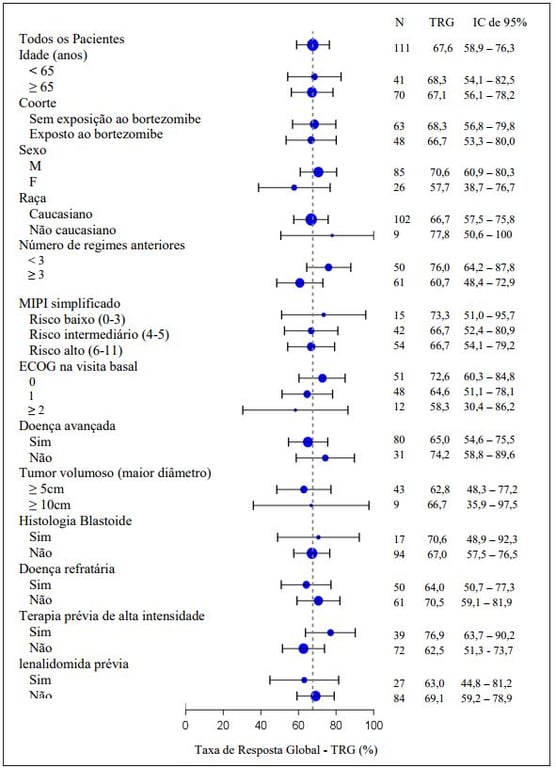
A resposta global para Ibrutinibe (substância ativa) foi independente de tratamento prévio, incluindo bortezomibe e lenalidomida ou risco/prognóstico subjacente, doença volumosa, gênero ou idade (figura a seguir).
Análise de subgrupo da Taxa de Resposta Global pela avaliação do investigador (Estudo PCYC- 1104-CA; 560 mg)
A segurança e eficácia de Ibrutinibe (substância ativa) foi demonstrada em um estudo de fase 3 randomizado, aberto, multicêntrico incluindo 280 pacientes com LCM que receberam pelo menos um tratamento prévio (Estudo MCL3001). Pacientes foram randomizados 1:1 para receber tanto Ibrutinibe (substância ativa) via oral, 560 mg uma vez ao dia em um ciclo de 21 dias, ou temsirolimo por via intravenosa na dose de 175 mg dos dias 1, 8, 15 do primeiro ciclo, seguido por 75mg nos dias 1, 8, 15 de cada ciclo de 21 dias subsequente. O tratamento em ambos os braços continuou até a progressão da doença ou toxicidade inaceitável.
A idade mediana foi de 68 anos (variação, 34 a 88 anos), 74% eram homens e 87% eram caucasianos. O tempo mediano desde o diagnóstico foi de 43 meses e o número mediano de tratamentos prévios foi 2 (variação, 1 a 9 tratamentos), incluindo 51% com quimioterapia prévia de alta dose, 18% com bortezomibe prévio, 5% com lenalidomida prévia e 24% com transplante de células tronco prévio. Na visita basal, 53% dos pacientes tiveram doença volumosa (? 5cm), 21% tinham pontuação de alto risco pelo Índice de Prognóstico Internacional simplificado (MIPI), 60% tinham doença extranodal e 54% tinham envolvimento de medula óssea na triagem.
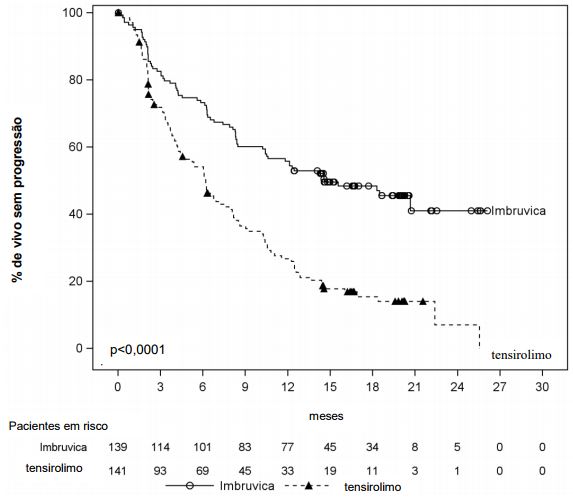
Sobrevida livre de progressão (SLP) conforme avalidado pelo CRI (Comitê de Revisão Independente) de acordo com os critérios revisados do IWG de linfoma não Hodgkin (LNH) demostraram uma redução estatisticamente significante de 57% no risco de morte ou progressão para pacientes no braço de Ibrutinibe (substância ativa). Resultados de eficácia para o estudo MCL3001 são demonstrados na tabela a seguir e a curva de Kaplan Meier para SLP na figura a seguir.
Resultados de Eficácia no estudo MCL3001
|
Desfecho |
01 Desfecho Ibrutinibe (substância ativa) N=139 |
Tensirolimo N=141 |
|
Sobrevida Livre de Progressãoa |
||
|
Número de eventos (%) |
73 (52,5) |
111 (78,7) |
|
Mediana de Sobrevida Livre de Progressão (IC de 95%), meses |
14,6 (10,4; NE) |
6,2 (4,2; 7,9) |
|
HR (IC de 95%) |
0,43 (0,32; 0,58) |
|
|
Taxa de Resposta Global (RC+RP) |
71,9% |
40,4% |
|
Valor de p |
p<0,0001 |
|
NE = Não estimável.
HR = hazard ratio (Razão de risco).
IC = intervalo de confiança.
RC = Resposta Completa.
RP = Resposta Parcial.
aAvaliado pelo CRI.
Uma proporção menor de pacientes tratados com Ibrutinibe (substância ativa) experimentaram uma piora clinicamente significativa de sintomas do linfoma versus tensirolimo (27% versus 52%) e o tempo para piora dos sintomas ocorreram mais lentamente com Ibrutinibe (substância ativa) versus tensirolimo (HR 0,27; p< 0,0001).
Curva Kaplan-Meier de Sobrevida Livre de Progressão (SLP) (População ITT) no estudo MCL3001
Leucemia linfocítica crônica/Linfoma linfocítico de pequenas células
A segurança e a eficácia de Ibrutinibe (substância ativa) em pacientes com LLC/LLPC que receberam no mínimo um tratamento anterior foram avaliadas em um estudo não controlado e num estudo controlado, randomizado.
Pacientes com LLC/LLPC que receberam pelo menos uma terapia anterior
No estudo aberto multicêntrico (PCYC-1102-CA), 51 pacientes que apresentavam LLC/LLPC foram tratados com Ibrutinibe (substância ativa), 420 mg uma vez ao dia até a progressão da doença ou até toxicidade inaceitável. A idade mediana dos pacientes foi de 68 anos (variação, 37 a 82 anos), tempo mediano desde o diagnóstico de 80 meses e número mediano de tratamentos anteriores de 4 ( variação, 1 a 12 tratamentos), incluindo 92% com uso prévio de análogo de nucleosídeo, 98% com uso prévio de rituximabe, 86% com uso prévio de alquilante, 39% com uso prévio de bendamustina e 20% com uso prévio de ofatumumabe. Na visita basal, 39,2% dos pacientes apresentaram estágio IV da classificação de Rai, 45% apresentaram doença volumosa (? 5 cm), 35% tiveram del 17p, 31% tiveram del 11q.
A taxa de resposta global foi avaliada pelo investigador de acordo com os critérios do Seminário Internacional de 2008 para LLC (IWCLL). Na duração mediana de acompanhamento de 16,4 meses, as respostas a Ibrutinibe (substância ativa) para os 51 pacientes são ilustradas na tabela a seguir.
Taxa de resposta global em pacientes com Leucemia Linfocítica Crônica tratados com 420 mg de Ibrutinibe (substância ativa) - Estudo PCYC-1102-CA (N=51)
|
Taxa de resposta global (RC+RP) (IC de 95% ) (%) |
78,4 (64,7; 88,7) |
|
RC (%) |
3,9 |
|
RP (%) |
74,5 |
|
Taxa de resposta global incluindo Resposta Parcial com linfocitose (RPL) (%) |
92,2 |
|
Mediana da duração da resposta (RC+RP) |
NA1 |
|
Tempo mediano para resposta inicial, meses (variação) |
1,8 (1,4-12,2) |
IC = intervalo de confiança.
RC = resposta completa.
RP = resposta parcial.
192,5% dos pacientes responsivos foram censurados (isto é, sem progressão e vivos) com acompanhamento mediano de 16,4 meses.
NA: não atingido.
Os dados de eficácia foram adicionalmente avaliados por um Comitê de Revisão Independente (CRI) usando o critério IWCLL e a taxa de resposta global obtida foi de 65% (95% CI: 50%, 78%), sendo todas respostas parciais. A duração da resposta variou de 4 meses a 24 + meses. A mediana da duração da resposta não foi alcançada.
Um estudo Fase 3 aberto, multicêntrico, radomizado de Ibrutinibe (substância ativa) versus ofatumumabe (PCYC- 1112-CA) foi conduzido em pacientes com LLC/LLPC. Pacientes (n=391) foram randomizados 1:1 para receber Ibrutinibe (substância ativa) 420 mg por dia até a progressão da doença ou toxicidade inaceitável, ou ofatumumabe por até 12 doses (300/2000 mg). Cinquenta e sete pacientes randomizados para ofatumumabe, após a progressão da doença, cruzaram para receber Ibrutinibe (substância ativa).
A idade mediana dos pacientes foi de 67 anos (variação, 30 a 88 anos), 68% eram homens e 90% eram Caucasianos. Todos os pacientes apresentaram um desempenho funcional “performance status” (ECOG), na visita basal, de 0 ou 1. A mediana de tempo desde o diagnóstico foi 91 meses e o número mediano de tratamentos anteriores foram dois (variação, 1 a 13 tratamentos). Na visita basal, 58% dos pacientes tinham no mínimo um tumor ?5 cm. Trinta e dois porcento dos pacientes tinham deleção 17p e 31% tinham deleção 11q.
A sobrevida livre de progressão (SLP) avaliada pelo Comitê de Revisão Independente (CRI) de acordo com os critérios do IWCLL indicaram uma redução estatisticamente significativa de 78% no risco de morte ou progressão para pacientes no braço de Ibrutinibe (substância ativa). Análise de sobrevida global (SG) demostrou uma redução estatiticamente significativa de 57% no risco de morte para pacientes no braço de Ibrutinibe (substância ativa). Os resultados de eficácia para o estudo PCYC-1112-CA estão demonstrados na tabela a seguir:
Resultados de eficácia em pacientes com Leucemia Linfocítica Crônica/Linfoma Linfocítico de Pequenas Células (Estudo PCYC-1112-CA)
|
Desfecho |
Ibrutinibe (substância ativa) N=195 |
Ofatumumabe N=196 |
|
Sobrevida Livre de Progressão |
||
|
Mediana de Sobrevida Livre de Progressão, meses |
Não alcançada |
8,1 |
|
HRf (ICg de 95%) |
||
|
Sobrevida Globala |
||
|
HR (IC de 95%) |
0,434 (0,238;0,789)b |
|
|
0,387 (0,216;0,695)c |
||
|
Taxa de Resposta Globald,e (%) |
42,6 |
4,1 |
|
Taxa de Resposta Global incluindo Resposta Parcial com Linfocitose (RPL)d (%) |
62,6 |
4,1 |
aSobrevida Global (SG) Mediana não alcançada por ambos os braços.
bPacientes randomizados para ofatumumabe que progrediram foram censurados ao iniciar Ibrutinibe (substância ativa) se aplicável.
cAnálise de sensibilidade na qual pacientes que cruzaram do braço de ofatumumabe não foram censurados na data da primeira dose de Ibrutinibe (substância ativa).
dPor CRI (Comite de revisão independente). Necessária repetição de exame de tomografia computadorizada para confirmar resposta.
eTodas as respostas parciais (RP) alcançadas. p < 0,0001 para taxa de resposta global.
fHR: Hazard Ratio (Razão de risco).
gIC: intervalo de confiança.
A eficácia foi semelhante em todos os subgrupos examinados, incluindo pacientes com e sem deleção 17p, um fator de estratificação pré-específicado (figura a seguir).
Análise de Subgrupo de Sobrevida Livre de Progressão pelo CRI (Estudo PCYC-1112-CA; 420 mg)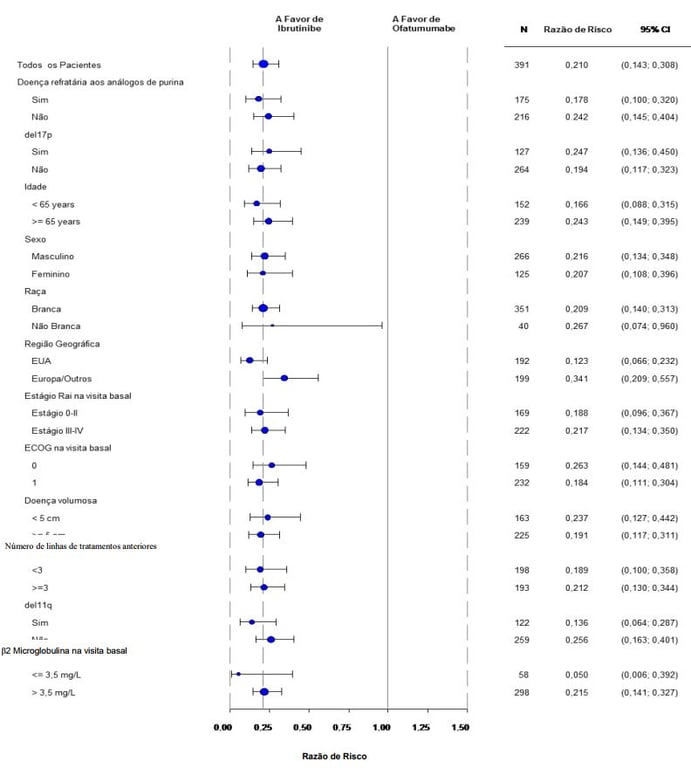
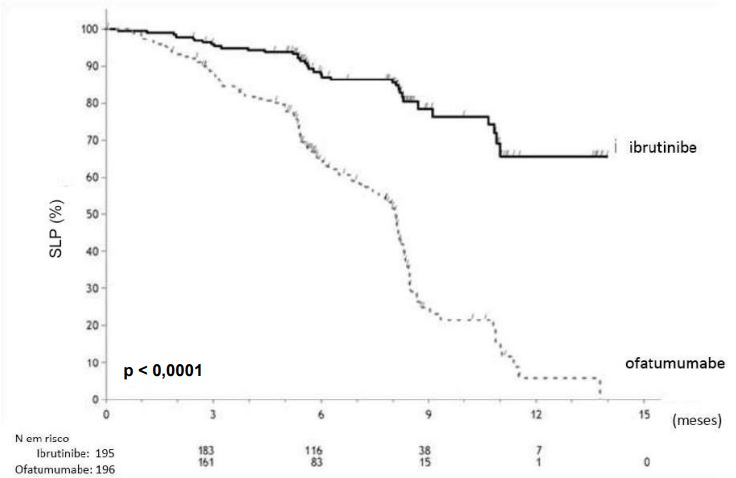
As curva Kaplan Meier para Sobrevida Livre de Progressão (SLP) e Sobrevida Global (SG) são demonstradas repectivamente nas figuras abaixo.
Curva Kaplan-Meier de Sobrevida Livre de Progressão (SLP) (População ITT) no estudo PCYC- 1112-CA
N: número de pacientes.
ITT: População com Intenção de Tratamento.
Curva Kaplan-Meier de Sobrevida Global (SG) (População ITT) no estudo PCYC-1112-CA
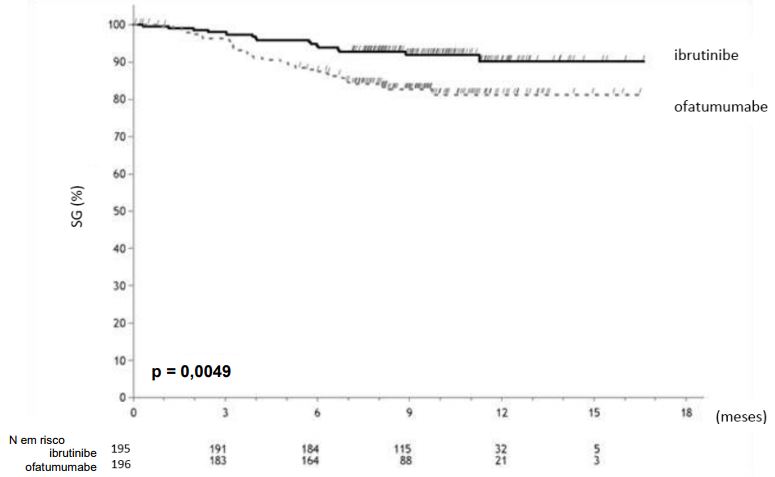
N: número de pacientes.
ITT: População com Intenção de Tratar.
Macroglobulinemia de Waldenström (MW)
A segurança e eficácia de Ibrutinibe (substância ativa) em MW (linfoma linfoplasmocítico excretor de IgM) foram avaliados em um estudo aberto, multicêntrico, braço único de 63 pacientes previamente tratados. A idade mediana foi 63 anos (variação, 44 a 86 anos), 76% eram homens, e 95% eram Caucasianos. Todos os pacientes tiveram classificação de performance ECOG na avaliação basal de 0 ou 1. O tempo mediano desde o diagnóstico foi 74 meses e o número mediano de tratamentos anteriores foi 2 (variação, 1 a 11 tratamentos). Na visita basal, o valor mediano de IgM sérica foi 3,5 g/dL (variação, 0,7 a 8,4g/dL) e 60% dos pacientes estavam anêmicos (hemoglobina ?11g/dL).
Ibrutinibe (substância ativa) foi administrado oralmente 420mg uma vez ao dia até a progressão da doença ou toxicidade inaceitável. O desfecho primário neste estudo foi a taxa de resposta global avaliada pelo investigador. A taxa de resposta global e a duração da resposta foram avaliadas usando critério adotado do Terceiro Workshop Internacional de Macroglobulinemia de Waldenström (Kimby E, 2006). As respostas de Ibrutinibe (substância ativa) são demostrada na tabela a seguir:
Taxa de Resposta Global (TRG) e Duração da Resposta (DR) baseado na avaliação do investigador em pacientes com WM
|
Total (N=63) |
|
|
TRG (%) |
87,3 |
|
IC de 95% (%) |
(76,5; 94,4) |
|
VGPR (%) |
14,3 |
|
RP (%) |
55,6 |
|
RM (%) |
17,5 |
| Duração da resposta mediana em meses (variação) |
NA (0,03+, 18,8+) |
IC = intervalo de confiança.
NA = não alcançado.
RM = resposta menor.
RP = resposta parcial.
RPMB = resposta parcial muito boa.
TRG = RM+RP+RPMB.
TRG: taxa de resposta global.
O tempo mediano para resposta foi 1,0 mês (variação 0,7 – 13,4 meses).
Os resultados de eficácia também foram avaliados por um Comitê de Revisão Independente, demostrando uma taxa de resposta global de 82,5%, com uma taxa de resposta parcial muito boa de 11% e uma taxa de resposta parcial de 51%.
Características farmacológicas
Propriedades farmacodinâmicas
Mecanismo de ação
O Ibrutinibe (substância ativa) é uma pequena molécula potente inibidora da tirosina quinase de Bruton (BTK). O Ibrutinibe (substância ativa) forma uma ligação covalente com um resíduo de cisteína (Cys-481) no sítio ativo da BTK, levando a inibição prolongada da atividade enzimática da BTK. A BTK, um membro da família Tec quinase, é uma importante molécula de sinalização do receptor antigênico da célula B (BCR) e vias de receptor de citocina. A via BCR está envolvida na patogênese de várias malignidades de células B, incluindo linfoma de células do manto (LCM), linfoma difuso de grandes células B, linfoma folicular e LLC de células B. O papel central da BTK na sinalização pelos receptores de superfície de células B resulta na ativação de vias necessárias para circulação de células B, quimiotaxia e adesão. Estudos pré-clínicos demonstraram que Ibrutinibe (substância ativa) inibe a proliferação e a sobrevida de células B malignas in vivo, bem como a migração celular e a adesão ao substrato in vitro.
De acordo com o estudo PCYC-1102-CA o tempo mediano para a resposta inicial ao Ibrutinibe (substância ativa) foi de 1,8 meses, variando de 1,4 meses a 12,2 meses.
Linfocitose
Após o início do tratamento, foi observado um aumento reversível nas contagens de linfócitos (isto é, aumento de ? 50% em relação ao valor basal e uma contagem absoluta > 5000/mcL), frequentemente associado à redução da linfoadenopatia, na maior parte dos pacientes (66%) que apresentavam LLC/LLPC tratados com Ibrutinibe (substância ativa). Este efeito também foi observado em alguns pacientes (35%) com LCM tratados com Ibrutinibe (substância ativa) como monoterapia. Esta linfocitose observada é um efeito farmacodinâmico e não deve ser considerado como progressão da doença na ausência de outros achados clínicos. Em ambos os tipos de doença, a linfocitose ocorre tipicamente durante o primeiro mês de terapia com Ibrutinibe (substância ativa) e tipicamente apresenta resolução no período mediano de 8 semanas em pacientes com LCM e de 14 semanas em pacientes com LLC/LLPC(variação 0,1 a 104 semanas).
Foi observado um grande aumento do número de linfócitos circulantes (por exemplo, > 400000/mcL) em alguns pacientes.
Linfocitose não foi observada em pacientes com MW tratados com Ibrutinibe (substância ativa).
Quando Ibrutinibe (substância ativa) foi administrado com quimioterapia, linfocitose não foi frequente (7% com Ibrutinibe (substância ativa) + BR versus 6% com placebo + BR).
Agregação plaquetária in vitro
Em um estudo in vitro, o Ibrutinibe (substância ativa) demonstrou inibição na agregação plaquetária induzida por colágeno em amostras de coortes de indivíduos com disfunção renal usando varfarina, ou com indivíduos sadios. A magnitude da inibição da agregação plaquetária induzida pelo colágeno no coorte de indivíduos com aspirina foi menos pronunciada uma vez que a agregação plaquetária induzida pelo colágeno já estava reduzida sem o Ibrutinibe (substância ativa). O Ibrutinibe (substância ativa) não mostrou inibição significativa da agregação plaquetária para os 4 agonistas adenosina difosfato (ADP), ácido araquidônico, ristocetina e do peptídeo-6 ativador do receptor de trombina (TRAP-6) em qualquer uma destes coortes de indivíduos ou indivíduos sadios.
Efeito no intervalo QT/QTc e eletrofisiologia cardíaca
O efeito do Ibrutinibe (substância ativa) no intervalo QTc foi avaliado em 20 homens e mulheres saudávies em um estudo de QT minucioso randomizado, duplo cego, com placebo e controles positivos. Numa dose supraterapeutica de 1680 mg, Ibrutinibe (substância ativa) não prolongou o intervalo QT a nenhuma extensão clinicamente relevante.
O limite superior mais alto do IC de 90% bicaudal para as diferenças médias ajustadas em relação ao basal entre Ibrutinibe (substância ativa) e placebo foi inferior a 10 ms. Observou-se no mesmo estudo, um encurtamento no intervalo QTc dependente da concentração [-5,3ms (IC de 90%: -9,4;-1,1) em um Cmáx de 719ng/mL seguido de uma dose supraterapêutica de 1680mg] que foi considerada clinicamente não relevante.
Propriedades Farmacocinéticas
Absorção
O Ibrutinibe (substância ativa) é rapidamente absorvido após a administração oral, com Tmáx mediano de 1 a 2 horas. A biodisponibilidade absoluta em condições de jejum (n = 8) foi 2,9% (IC 90% = 2,1 – 3,9) e duplicou quando combinado com uma refeição. A farmacocinética de Ibrutinibe (substância ativa) não difere significativamente em pacientes que apresentam diferentes malignidades de células B. A exposição ao Ibrutinibe (substância ativa) aumenta com o incremento de doses até 840 mg. A AUC no estado de equilíbrio observada em pacientes com 560 mg é (média ± desvio) 953 ± 705 ng·h/mL.
A administração de Ibrutinibe (substância ativa) em condições de jejum resulta em aproximadamente 60% da exposição (AUCúltima) quando comparado com 30 minutos antes, 30 minutos depois (condição de alimentação) ou duas horas após uma café da manhã hiperlipídico. Em um estudo clínico (Estudo CLL1001) em indivíduos saudáveis, a média da exposição em 3 diferentes condições de alimentação ( Ibrutinibe (substância ativa) 30 minutos após ou 30 minutos antes ou 2 horas após um café da manhã hiperlipídico), foram similares àquelas obervadas em pacientes com LLC no estudo 1102 (Ibrutinibe (substância ativa) foi administrado pelo menos 30 minutos antes ou 2 horas após a refeição). Neste mesmo estudo, os dados disponíveis de segurança permitem o uso seguro de Ibrutinibe (substância ativa) em qualquer condição de alimentação.
Distribuição
A ligação reversível de Ibrutinibe (substância ativa) às proteínas plasmáticas humanas in vitro foi de 97,3%, sem dependência da concentração na variação de 50 a 1000 ng/mL. O volume de distribuição (Vd) foi 683 L e o volume de distribuição aparente em estado de equilíbrio (Vd,ss/F) é de aproximadamente 10000 L.
Metabolismo
O Ibrutinibe (substância ativa) é metabolizado primariamente pelo citocromo P450, CYP3A4/5, para produzir um metabólito di-hidrodiol com atividade inibidora de BTK aproximadamente 15 vezes inferior à de Ibrutinibe (substância ativa). A exposição sistêmica em estado estável ao metabólito di-hidrodiol é comparável à do medicamento precursor.
Estudos in vitro indicaram que o envolvimento de CYP2D6 no metabolismo oxidante de Ibrutinibe (substância ativa) é < 2%. Além disso, como parte do estudo de equilíbrio de massa em humanos, os pacientes genotipados como metabolizadores fracos para CYP2D6 apresentaram perfil farmacocinético semelhante ao dos metabolizadores intensos. Portanto, não são necessárias precauções em pacientes com diferentes genótipos de CYP2D6.
Eliminação
A depuração intravenosa foi 62 e 76 L/h em condições de jejum e de alimentação, respectivamente. Em linha com o alto efeito de primeria passagem, a depuração aparente oral é aproximadamente 2000 e 1000 L/h em condições de jejum e de alimentação, respectivamente. A meia-vida de Ibrutinibe (substância ativa) é de 4 a 6 horas.
Após uma administração oral única de [14C]-Ibrutinibe (substância ativa) radiomarcado em indivíduos saudáveis, aproximadamente 90% da radioatividade foram excretados no período de 168 horas, com a maior parte (80%) excretada nas fezes e menos de 10% na urina. O Ibrutinibe (substância ativa) não metabolizado totalizou aproximadamente 1% do produto de excreção radiomarcado nas fezes e nada na urina, com o restante da dose sendo metabólitos.
Pacientes idosos (? 65 anos de idade)
A farmacocinética da população indicou que em pacientes mais idosos (67 a 81 anos), é previsto um aumento 14% maior na exposição ao Ibrutinibe (substância ativa). Não se justifica ajuste de dose por idade.
Pacientes pediátricos (? 18 anos de idade)
Não foram realizados estudos farmacocinéticos com Ibrutinibe (substância ativa) em pacientes com menos de 18 anos de idade.
Sexo
Os dados de farmacocinética da população indicaram que o sexo não influencia significativamente a eliminação de Ibrutinibe (substância ativa) da circulação.
Insuficiência renal
O Ibrutinibe (substância ativa) possui depuração renal mínima; a excreção urinária dos metabólitos é <10% da dose. Não foram realizados estudos clínicos específicos até o momento em pacientes com insufuciência renal.
Não é necessário ajuste de dose para pacientes que apresentam insufuciência renal leve ou moderada (depuração de creatinina maior do que 30 mL/min). Não há dados em pacientes com insufuciência renal grave ou pacientes em diálise.
Insuficiência hepática
O Ibrutinibe (substância ativa) é metabolizado no fígado. Um ensaio de insuficiência hepática foi realizado em pacientes sem câncer, administrada uma única dose de 140 mg de Ibrutinibe (substância ativa) , em condições de jejum. A AUCúltima de Ibrutinibe (substância ativa) aumentou 2,7-, 8,2- e 9,8- vezes em pacientes com insuficiência hepática leve (n=6; Child-Pugh classe A), moderada (n=10; Child-Pugh classe B) e severa (n=8; Child-Pugh classe C), respectivamente.
As frações livres de Ibrutinibe (substância ativa) também aumentaram com o grau de insuficiência, com 3,0; 3,8 e 4,8% em pacientes com insuficiência hepática leve, moderada e severa, respectivamente, em comparação a 3,3% no plasma proveniente de controles saudáveis pareados dentro deste estudo. O aumento correspondente na exposição ao Ibrutinibe (substância ativa) não ligado (AUCnão ligado, última) é estimado para ser 4,1-, 9,8- e 13- vezes em pacientes com insuficiência hepática leve, moderada e severa, respectivamente.
Cuidados de Armazenamento
Conservar em temperatura ambiente (entre 15°C e 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original. Após aberto, válido por 45 dias.
Aspecto físico
Cápsula de gelatina dura, branca e opaca, com “ibr 140mg” gravado em preto, contendo pó branco a quase branco.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS - 1.1236.3412
Farm. Resp.:
Marcos R. Pereira - CRF/SP n° 12.304
Registrado por:
JANSSEN-CILAG FARMACÊUTICA LTDA.
Avenida Presidente Juscelino Kubitschek, 2041, São Paulo – SP
CNPJ 51.780.468/0001-87
Fabricado por:
Catalent Clinical Trials Supplies LLC
Kansas City - EUA
Embalado por:
AndersonBrecon, Inc.
Rockford - EUA
Importado por:
Janssen-Cilag Farmacêutica Ltda.
Rodovia Presidente Dutra, km 154
São José dos Campos -SP
CNPJ 51.780.468/0002-68
Venda sob Prescrição Médica.
Comparar preços de remédios e medicamentos no CliqueFarma é rápido e simples.
O CliqueFarma, é uma ferramenta para comparativo de preços de produtos farmacêuticos. Não comercializamos, não indicamos, não receitamos, nenhum tipo de medicamento essa função cabe exclusivamente a médicos e farmacêuticos. Não consuma qualquer tipo de medicamento sem consultar seu médico.
SE PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO. PROCURE UM MÉDICO E O FARMACÊUTICO. LEIA A BULA.
Conheça nossos Termos de Uso