Comparamos o preço de Certican 0,50 Mg 60 Comprimidos, veja o menor preço

R$ 1.761,00
RReferência
15
ofertasMelhores preços a partir de R$ 1.761,00 até R$ 2.310,35
Menor preço
vendido por Maranata Medicamentos
economize
23.78%
R$ 1.761,00
vendido por OncoExpresso Medicamentos
economize
20.36%
R$ 1.840,00
vendido por Life Medicamentos
economize
19.59%
R$ 1.857,70
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Farma Visa
economize
19.59%
R$ 1.857,70
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Imune Farma Medicamentos Especiais
economize
19.59%
R$ 1.857,80
vendido por Farma Ame
economize
19.59%
R$ 1.857,85
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Remédio Já
economize
19.54%
R$ 1.859,00
vendido por Saúde Farma Medicamentos
economize
19.36%
R$ 1.863,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
vendido por Farma Silva
economize
17.12%
R$ 1.914,77
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Pague Menos
economize
7.50%
R$ 2.136,99
vendido por Facilita Medicamentos
economize
4.15%
R$ 2.214,41
vendido por Drogaria Dinâmica
economize
3.82%
R$ 2.222,10
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Mundial Farma
economize
3.56%
R$ 2.228,00
vendido por Onco Express Medicamentos Especiais e Oncológicos
economize
3.10%
R$ 2.238,72
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
vendido por Fast Medicamentos
R$ 2.310,35
Para que serve
Certican é utilizado para ajudar na prevenção da rejeição de rim, coração ou fígado transplantados.
Certican deve ser utilizado juntamente com outros medicamentos imunossupressores, como a ciclosporina para microemulsão para transplante de rim e de coração, ou tacrolimo para transplante de fígado, e corticosteroides.
Como o Certican funciona?
Certican pertence ao grupo de medicamentos conhecidos como imunossupressores (os imunossupressores reduzem a atividade do sistema de defesa do seu organismo e são essenciais para ajudar na prevenção da rejeição dos órgãos transplantados).
Contraindicação
Se você é alérgico (hipersensível) ao everolimo, sirolimo ou a qualquer um dos componentes de Certican.
Os componentes contidos em cada comprimido estão listados no início desta bula.
Avise ao seu médico caso você suspeite que já tenha apresentado reação alérgica a qualquer um dos componentes no passado.
Como usar
Seu médico decidirá a dose exata e quando você deverá tomar Certican.
Siga as instruções do seu médico cuidadosamente e nunca altere a dose por você mesmo.
Não exceda a dose recomendada.
Certican deve ser usado apenas por administração oral.
Quando e como tomar de Certican
Certican pode ser tomado com ou sem alimentos, mas deve ser sempre tomado com alimentos ou sempre sem alimentos. Não tome Certican com toranja ou suco de toranja.
Quanto tomar de Certican em casos de transplantes do coração e rim
A dose diária recomendada geral é de 0,75 mg duas vezes ao dia, de manhã e à noite, junto com a ciclosporina para microemulsão.
A primeira dose de Certican deve ser administrada o mais rápido possível após o transplante.
Quanto tomar de Certican em casos de transplante de fígado
A dose diária recomendada, em geral, é de 1 mg de Certican, duas vezes ao dia, de manhã e à noite, junto com tacrolimo.
A primeira dose de Certican deve ser administrada aproximadamente quatro semanas após o transplante.
Sua dose poderá ser ajustada dependendo do nível de Certican no seu sangue e sinais clínicos. Seu médico precisará realizar testes sanguíneos regularmente para medir os níveis de Certican.
Por quanto tempo tomar Certican
O tratamento deve continuar enquanto você precisar de imunossupressão para prevenir a rejeição ao órgão transplantado.
Se você parar de tomar Certican
A interrupção do seu tratamento com Certican pode aumentar a possibilidade da rejeição de seu órgão transplantado.
Não pare de tomar o medicamento a menos que seu médico o diga para fazê-lo.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
Precauções
Certican somente será receitado para você por um médico com experiência em transplante.
Siga as instruções do seu médico cuidadosamente. Elas podem diferir das informações gerais contidas nesta bula.
Tome cuidado especial com Certican se você desenvolver qualquer um dos seguintes sintomas
- Inchaço da face, lábios, garganta ou língua, ou repentina dificuldade em engolir ou respirar;
- Persistência ou piora dos sintomas pulmonares/respiratórios, como tosse, respiração dificultada ou ofegante;
- Hematomas na pele sem causa aparente;
- Dor, hérnia, calor incomum, inchaço ou saída de líquido do local da cirurgia;
- Redução brusca em sua produção de urina, especialmente se acompanhada de dor no local do rim transplantado.
Se apresentar estes sintomas, informe ao seu médico imediatamente.
Medicamentos que suprimem o sistema imunológico, como o Certican, reduzem a capacidade do seu organismo de combater infecções. É aconselhável consultar o seu médico ou centro de transplante em caso de febre, mal-estar ou sintomas locais, tais como tosse, sensação de ardor ao urinar, que são graves ou persistentes durante vários dias.
Medicamentos que suprimem o sistema imunológico, como o Certican, aumentam o risco de desenvolvimento de câncer, particularmente de pele e sistema linfático. Portanto, você deve reduzir a exposição à luz solar e luz ultravioleta (UV) com o uso de roupas protetoras apropriadas e uso frequente de protetor solar com alto fator de proteção.
Se você já apresentou algum problema de fígado ou teve alguma doença que possa ter afetado seu fígado, por favor, informe seu médico, pois pode haver necessidade de alteração da dose de Certican que você tenha que tomar.
Muitos medicamentos podem interagir com Certican. Por favor, informe ao seu médico sobre outros medicamentos que esteja usando, especialmente se você está tomando quaisquer medicamentos que contenham rifampicina, rifabutina, cetoconazol, itraconazol, voriconazol, claritromicina, telitromicina ou ritonavir. Pode ser necessário modificar a dose de Certican.
Se você precisa receber uma vacina, peça antes orientação ao seu médico.
Se você apresentar sintomas respiratórios (por exemplo: tosse, respiração dificultada e ofegante), por favor, informe ao seu médico. Seu médico decidirá se você precisa continuar com Certican e/ou se você precisa receber outros medicamentos para resolver esta condição.
Certican pode reduzir a qualidade dos espermatozoides em homens, reduzindo a capacidade de ter filhos. O efeito geralmente é reversível. Pacientes do sexo masculino que desejam ter filhos devem discutir o tratamento com o médico.
Em função da pouca experiência clínica com agentes imunossupressores desta categoria (everolimo e sirolimo), pacientes de ambos os sexos devem usar métodos contraceptivos até que informações mais conclusivas possam ser obtidas.
Monitoramento durante o tratamento com Certican
Exames regulares de sangue e urina são essenciais para o seu médico avaliar o bom funcionamento do órgão transplantado, detectar possíveis efeitos indesejados do medicamento e adaptar as doses de seus medicamentos para obter o melhor tratamento.
Os exames de sangue permitem ao médico medir os níveis dos medicamentos (everolimo, ciclosporina e tacrolimo), verificar sua função renal e os níveis de açúcar e de colesterol em seu sangue.
A análise das proteínas em uma amostra de urina também ajuda o médico a avaliar a atividade dos rins.
Reações Adversas
Como todos os medicamentos, Certican apresenta reações adversas, embora nem todas as pessoas apresentem.
Entretanto, como Certican é administrado com outros medicamentos, as reações adversas nem sempre podem ser atribuídas diretamente ao Certican.
Algumas reações adversas podem ser graves
Inflamação dos pulmões
Informe ao seu médico imediatamente se tiver persistência/piora dos sintomas pulmonares/respiratórios, como tosse, respiração dificultada ou ofegante. Isso pode indicar que você tem inflamação pulmonar, que pode ser fatal. Seu médico poderá precisar interromper o tratamento com Certican ou adicionar outro medicamento para ajudar com essa reação adversa.
Infecção
Certican pode aumentar o risco de adquirir infecções (por exemplo, infecções respiratórias, infecções urinárias, infecções virais ou fúngicas em geral). Estas infecções podem ser graves e até fatais. Informe ao seu médico imediatamente se você tiver aumento de temperatura, tosse, calafrios ou outros sinais de uma infecção, pois você pode precisar de tratamento urgente.
Angioedema
Certican pode causar angioedema que, tipicamente, aparece como um inchaço súbito da face, lábios, língua ou garganta. Informe ao seu médico imediatamente, pois isso pode levar a dificuldades de deglutição e respiração, que podem ser fatais.
Microangiopatia trombótica
É uma doença pós-transplante que pode ocorrer com Certican. Provoca uma redução brusca do número de plaquetas no sangue. As plaquetas ajudam a parar o sangramento. Você deve informar o seu médico imediatamente se notar hematomas espontâneos ou sangrar sem motivo aparente.
Trombose do enxerto renal
É a obstrução repentina dos vasos sanguíneos que suprem o rim transplantado. Normalmente, ocorre dentro do primeiro mês após o transplante. Informe ao seu médico imediatamente se você tiver uma queda significativa na produção de urina, especialmente se acompanhada de dor no local do rim transplantado.
Se você apresentar qualquer uma destas reações, informe ao seu médico imediatamente.
Algumas reações adversas são muito comuns (ocorrem em mais de 10% dos pacientes que utilizam este medicamento)
- Infecções (viral, bacteriana e fúngica);
- Infecções do trato respiratório inferior, tais como infecções pulmonares e pneumonia;
- Infecções do trato respiratório superior, tais como inflamação na faringe e resfriado comum;
- Infecções no trato urinário;
- Anemia (contagem reduzida de células vermelhas no sangue);
- Contagem reduzida de plaquetas no sangue, que pode levar a hemorragia e/ou hematomas sob a pele;
- Níveis muito altos de algumas gorduras (lipídeos, colesterol e triglicérides) no sangue;
- Níveis reduzidos de potássio no sangue;
- Baixos níveis de glóbulos brancos (aumento do risco de infecções);
- Problemas para dormir (insônia);
- Ansiedade;
- Dor de cabeça;
- Acúmulo de fluido na bolsa ao redor do coração, que, quando grave, pode diminuir a habilidade do coração em bombear o sangue;
- Trombose venosa (bloqueio de uma veia principal por um coágulo sanguíneo);
- Acúmulo de fluido na cavidade pulmonar/torácica, que, quando grave, pode levar a falta de ar;
- Tosse;
- Falta de ar;
- Diarreia;
- Náusea;
- Vômito;
- Início de diabetes (nível elevado de açúcar no sangue);
- Pressão arterial alta;
- Dor abdominal;
- Dor generalizada;
- Edema (acúmulo de líquido nos tecidos);
- Cicatrização anormal de feridas;
- Febre.
Se você tiver dúvidas sobre alguma destas reações, informe ao seu médico.
Algumas reações adversas são comuns (ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento)
- Envenenamento do sangue;
- Infecção da ferida;
- Tumores benignos e cânceres;
- Câncer do tecido linfático (linfoma, desordem linfoproliferativa pós-transplantes);
- Batimento cardíaco acelerado;
- Hemorragias nasais;
- Dor nas articulações;
- Dor muscular;
- Dor na cavidade oral, como a garganta;
- Acne;
- Trombose do enxerto renal (obstrução súbita dos vasos sanguíneos que nutrem o rim transplantado que pode resultar em perda do enxerto);
- Redução simultânea dos glóbulos brancos e vermelhos e das plaquetas (os sintomas podem incluir fraqueza, hematomas e infecções frequentes);
- Cistos contendo líquido linfático;
- Inchaço na face, lábios, boca, língua ou garganta geralmente associadas com erupção cutânea e coceira;
- Inflamação do pâncreas (os sintomas podem incluir dor grave na parte superior do estômago, vômitos e perda de apetite);
- Feridas na boca;
- Proteína na urina;
- Distúrbio no rim;
- Impotência;
- Hérnia no local da cirurgia;
- Resultados anormais de testes no fígado;
- Rash (erupção cutânea).
Se você tiver dúvidas sobre alguma destas reações, informe ao seu médico.
Algumas reações adversas são incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento)
- Hemólise (destruição de glóbulos vermelhos);
- Inflamação dos pulmões (os sintomas podem incluir tosse, respiração dificultada e ofegante);
- Doença hepática geralmente com mal-estar;
- Icterícia (amarelamento da pele e olhos com urina escurecida);
- Câncer de pele;
- Diminuição do número de espermatozoides no esperma (diminui a probabilidade dos homens serem capazes de ter filhos).
Se você tiver dúvidas sobre alguma destas reações, informe ao seu médico.
Outras reações adversas (ocorrem em um pequeno número de pessoas, mas sua exata frequência é desconhecida)
- Acúmulo anormal de proteínas nos pulmões (os sintomas podem incluir tosse seca persistente, fadiga, dificuldade em respirar);
- Inflamação dos vasos sanguíneos (erupção localizada na pele);
- Grave erupção cutânea com inchaço da pele.
Se você tiver dúvidas sobre alguma destas reações, informe ao seu médico.
Além disso, podem ocorrer reações adversas inesperadas, como resultados laboratoriais anormais, incluindo testes de função renal. Durante o tratamento com Certican, seu médico irá solicitar exames de sangue para monitorar qualquer alteração.
Se você perceber qualquer reação adversa que não esteja mencionada nesta bula ou esteja relacionada com as reações listadas, por favor, avise seu médico.
Atenção: este produto é um medicamento que possui nova indicação no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidas. Nesse caso, informe seu médico.
População Especial
Pacientes idosos (pacientes com 65 anos ou mais)
A experiência do uso de Certican em pacientes idosos é limitada.
Crianças e adolescentes
A experiência do uso de Certican em crianças e adolescentes é limitada.
Gravidez e lactação
Peça orientações para seu médico ou farmacêutico antes de tomar qualquer medicamento.
- Certican não deve ser utilizado durante a gravidez, exceto quando indicado claramente como necessário pelo seu médico. Se você estiver grávida ou acha que pode estar grávida, consulte seu médico, ele analisará o risco potencial de tomar Certican durante a gravidez;
- A amamentação deve ser descontinuada em pacientes tomando Certican.
?Mulheres em idade fértil
Você deve usar um método contraceptivo efetivo durante o tratamento com Certican e também durante oito semanas após a interrupção do tratamento. Se você acha que está grávida ou se está pensando em engravidar, consulte seu médico antes de tomar Certican.
Fertilidade
Certican pode ter um impacto na fertilidade masculina.
Habilidade de dirigir e/ou operar máquinas
Não foram realizados estudos específicos dos efeitos de Certican sobre a habilidade de dirigir e operar máquinas. Não se espera que Certican afete a habilidade de dirigir veículos ou operar máquinas.
Informações sobre alguns dos ingredientes de Certican
Caso você tenha diagnóstico de intolerância a determinados açúcares (glicose, galactose, lactose), informe ao seu médico antes de tomar Certican. Certican contém lactose.
Composição
Cada comprimido contém 0,5 mg, 0,75 mg ou 1,00 mg de everolimo.
Excipientes: butil-hidroxitolueno, estearato de magnésio, lactose monoidratada, hipromelose, crospovidona e lactose.
Superdosagem
Se você tomar mais comprimidos do que deveria ou se acidentalmente alguém mais tomou o seu medicamento, imediatamente avise o seu médico ou procure um hospital.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
O Everolimo (substância ativa) é preferencialmente metabolizado pela CYP3A4 no fígado e em alguma extensão na parede intestinal. Adicionalmente, é um substrato para a bomba de efluxo multi-droga, glicoproteína-P (PgP). Portanto, a absorção e subsequente eliminação do Everolimo (substância ativa) absorvido sistemicamente podem ser influenciadas por fármacos que afetam a CYP3A4 e/ou a PgP.
Interações observadas resultantes do uso concomitante não sendo recomendado
Rifampicina (indutor da CYP3A4)
O pré-tratamento de voluntários sadios com dose múltipla de rifampicina seguida de uma dose única de Everolimo (substância ativa) elevou o clearance (depuração) de Everolimo (substância ativa) em aproximadamente 3 vezes e diminuiu a Cmax em 58% e a ASC em 63%. A combinação com rifampicina não é recomendada.
Cetoconazol (inibidor da CYP3A4)
O pré-tratamento de voluntários sadios com doses múltiplas de cetoconazol seguido de uma dose única de Everolimo (substância ativa) aumentou o Cmax do Everolimo (substância ativa) em 3,9 vezes e a ASC em 15,0 vezes.
Interações antecipadas resultantes do uso concomitante não sendo recomendado
Fortes inibidores, indutores da CYP3A4
Não é recomendado o tratamento concomitante com fortes inibidores e/ou indutores do CYP3A4 (por exemplo, itraconazol, voriconazol, claritromicina, telitromicina, ritonavir e/ou rifampicina, rifabutina).
Interações observadas a serem consideradas
Ciclosporina (inibidor da CYP3A4/PgP)
A biodisponibilidade do Everolimo (substância ativa) foi aumentada significativamente pela coadministração de ciclosporina. Em um estudo de dose única em voluntários sadios, a ciclosporina para microemulsão aumentou a ASC de Everolimo (substância ativa) em 168% (faixa, 46% a 365%) e a Cmax em 82% (faixa, 25% a 158%) comparado com a administração de Everolimo (substância ativa) sozinho. Pode ser necessário ajuste de dose de Everolimo (substância ativa) se a dose de ciclosporina for alterada.
Eritromicina (inibidor da CYP3A4)
O pré-tratamento de indivíduos sadios com doses múltiplas de eritromicina seguido por uma dose única de Everolimo (substância ativa) aumentou o Cmax do Everolimo (substância ativa) em 2,0 vezes e ASC em 4,4 vezes.
Verapamil (inibidor da CYP3A4)
O pré-tratamento de indivíduos sadios com doses múltiplas de verapamil seguido por uma dose única de Everolimo (substância ativa) aumentou o Cmax do Everolimo (substância ativa) em 2,3 vezes e ASC em 3,5 vezes.
Ciclosporina (inibidor CYP3A4/PgP)
O Everolimo (substância ativa) teve uma influência clínica menor na farmacocinética da ciclosporina em pacientes receptores de transplante renal e cardíaco recebendo ciclosporina para microemulsão.
Octreotida
A coadministração de Everolimo (substância ativa) com octreotida depositada aumentou o Cmin de octreotida numa relação da média geométrica (Everolimo (substância ativa)/placebo) de 1,47 vezes.
Atorvastatina (substrato da CYP3A4) e pravastatina (substrato da PgP)
A administração de dose única de Everolimo (substância ativa) tanto com atorvastatina quanto com pravastatina em voluntários sadios não influenciou a farmacocinética da atorvastatina, da pravastatina e do Everolimo (substância ativa), assim como a biorreatividade total da HMG-CoA redutase no plasma a uma extensão clinicamente relevante. Entretanto, estes resultados não podem ser extrapolados aos outros inibidores da HMG-CoA redutase.
Os pacientes devem ser monitorados para o desenvolvimento de rabdomiólise e outras reações adversas descritas nas bulas dos inibidores da HMG-CoA redutase.
Midazolam (substrato da CYP3A4A)
Em um estudo de interação cruzada de fármacos, de dois períodos e a uma sequência fixa, 25 voluntários sadios receberam uma dose oral única de 4 mg de midazolam no período 1. No período 2, eles receberam 10 mg de Everolimo (substância ativa), uma vez ao dia por 5 dias e uma dose única de 4 mg de midazolam com a última dose de Everolimo (substância ativa). A Cmax do midazolam aumentou 1,25 vezes (IC 90% 1,14 – 1,37) e a ASCinf aumentou 1,30 vezes (1,22 – 1,39). A meia-vida do midazolam manteve-se inalterada. Esse estudo indicou que o Everolimo (substância ativa) é um fraco inibidor da CYP3A4.
Interações antecipadas a serem consideradas
Indutores moderados de CYP3A4
Indutores da CYP3A4 podem aumentar o metabolismo de Everolimo (substância ativa) e reduzir os níveis sanguíneos de Everolimo (substância ativa) (por exemplo: erva-de-São-João (Hypericum perforatum); anticonvulsivantes: carbamazepina, fenobarbital, fenitoína; medicamentos anti-HIV (por exemplo, efavirenz, nevirapina)).
Inibidores moderados de CYP3A4
Inibidores moderados do CYP3A4 e PgP podem elevar os níveis sanguíneos de Everolimo (substância ativa) (por exemplo, agentes antifúngicos: fluconazol; bloqueadores de canal de cálcio: nicardipino, diltiazem; inibidores de proteases: nelfinavir, indinavir, amprenavir).
Inibidores da PgP
Inibidores da PgP podem diminuir o efluxo de Everolimo (substância ativa) das células intestinais e aumentar as concentrações sanguíneas de Everolimo (substância ativa).
Substratos de CYP3A4 e CYP2D6
In vitro, o Everolimo (substância ativa) foi um inibidor competitivo da CYP3A4 e da CYP2D6, aumentando potencialmente as concentrações dos medicamentos eliminados por estas enzimas. Portanto, deve-se ter cautela na coadministração de Everolimo (substância ativa) com substratos da CYP3A4 e CYP2D6 com índice terapêutico estreito. Todos os estudos de interação in vivo foram conduzidos sem a administração concomitante de ciclosporina.
Vacinação
Imunossupressores podem afetar a resposta a vacinações e, portanto, vacinações durante o tratamento com Everolimo (substância ativa) podem ser menos eficazes. Vacinas com vírus vivos devem ser evitadas.
Interação Alimentícia
Toranja e suco de toranja afetam a atividade do citocromo P450 e da PgP e devem, portanto, ser evitados.
Ação da Substância
Resultados da eficácia
Transplante renal
O Everolimo (substância ativa) em doses fixas de 1,5mg/dia e 3mg/dia em associação com doses padrões de ciclosporina para microemulsão e corticosteroides foi estudado em dois estudos clínicos de fase III em receptores de transplante renal “de novo” (B201 e B251). Como comparador foi utilizado o micofenolato de mofetila (MMF) na dose de 1 g duas vezes ao dia. Os desfechos coprimários compostos foram falha de eficácia (rejeição aguda confirmada por biópsia, perda do enxerto, morte ou perda de acompanhamento) no mês 6, e perda do enxerto, morte ou perda do acompanhamento com 12 meses. Em geral, Everolimo (substância ativa) não se mostrou inferior ao MMF nestes estudos. A incidência de rejeição aguda confirmada por biópsia no mês 6, no estudo B201 foi 21,6%, 18,2% e 23,5% para os grupos de Everolimo (substância ativa) 1,5mg/dia, Everolimo (substância ativa) 3,0mg/dia e MMF, respectivamente. No estudo B251, as incidências foram 17,1%, 20,1% e 23,5% para grupos Everolimo (substância ativa) 1,5mg/dia, Everolimo (substância ativa) 3mg/dia e MMF, respectivamente.
Foi observada com maior frequência redução da função do aloenxerto com aumento da creatinina sérica em pacientes utilizando combinação de Everolimo (substância ativa) com doses plenas de ciclosporina para microemulsão do que em pacientes tratados com MMF. Este efeito sugere que o Everolimo (substância ativa) aumenta a nefrotoxicidade da ciclosporina. A análise da farmacodinâmica-concentração do medicamento mostrou que a função renal pode ser melhorada com a exposição reduzida à ciclosporina mantendo a eficácia enquanto a concentração sanguínea mínima de Everolimo (substância ativa) foi mantida acima de 3 ng/mL. Este conceito foi posteriormente confirmado em dois estudos adicionais fase III (A2306 e A2307, incluindo 237 e 256 pacientes respectivamente) que avaliaram a eficácia e segurança de Everolimo (substância ativa) 1,5 e 3mg por dia (dose inicial; a dose subsequente foi baseada na concentração mínima (C0) pretendida de ?3 ng/mL) em associação com uma exposição reduzida à ciclosporina. Nos dois estudos, houve melhora da função renal sem o comprometimento da eficácia. No entanto, nestes estudos, não houve braço comparativo sem Everolimo (substância ativa).
O estudo A2309 fase III, multicêntrico, randomizado, aberto, controlado, foi realizado com 833 receptores de transplante renal “de novo”, que foram randomizados para um dos dois regimes de doses diferentes de Everolimo (substância ativa), em combinação com dose reduzida de ciclosporina ou um regime padrão de micofenolato de sódio (MPA) + ciclosporina, e tratados por 12 meses. Todos os pacientes receberam terapia de indução com basiliximabe no pré-transplante e no dia 4 pós-transplante. Esteroides podiam ser administrados conforme necessários após o transplante.
As doses iniciais dos dois grupos de Everolimo (substância ativa) foram 1,5mg/d e 3mg, duas vezes ao dia, que foram, modificadas a partir do dia 5 para manter os níveis sanguíneos de Everolimo (substância ativa) no alvo de 3 a 8 ng/mL e 6 a 12 ng/mL, respectivamente.
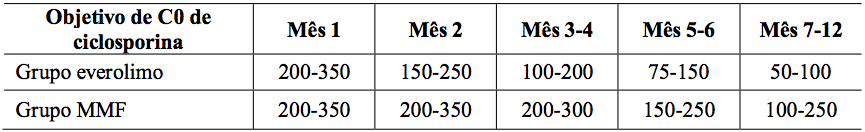
A dose de micofenolato de sódio foi de 1,44 g/d. As doses de ciclosporina foram adaptadas para manter os níveis sanguíneos mínimos no intervalo alvo, como mostrado na Tabela 1. Os valores reais medidos das concentrações sanguíneas de Everolimo (substância ativa) e ciclosporina (Co e C2) são mostrados na Tabela 2.
Embora o regime de dose mais alta com Everolimo (substância ativa) tenha sido tão eficaz quanto o regime com dose mais baixa, a segurança em geral foi pior e, por isso, o regime de dose maior não é recomendado.
O regime recomendado é o de dose mais baixa de Everolimo (substância ativa).
Tabela 1: Estudo A2309: Intervalo do nível mínimo alvo de ciclosporina no sangue:
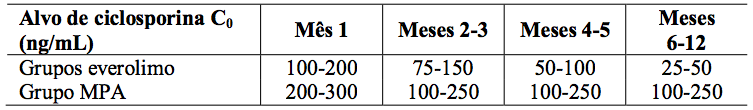
Tabela 2: Estudo A2309: Níveis sanguíneos mínimos medidos de ciclosporina e Everolimo (substância ativa):
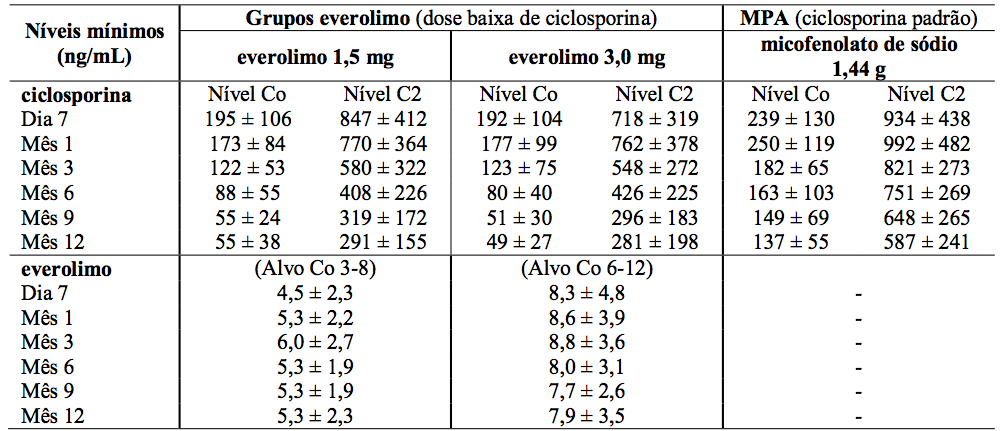
Os valores são a média ± DP das medidas dos valores de Co = nível, C2 = valores de 2 horas pós-dose.
O desfecho primário de eficácia foi uma variável combinada de falha (rejeição aguda comprovada por biópsia, perda do enxerto, morte ou perda de acompanhamento). O resultado é demonstrado na Tabela 3.
Tabela 3: Estudo A2309: Desfecho composto e individual de eficácia nos meses 6 e 12 (incidência em população ITT):
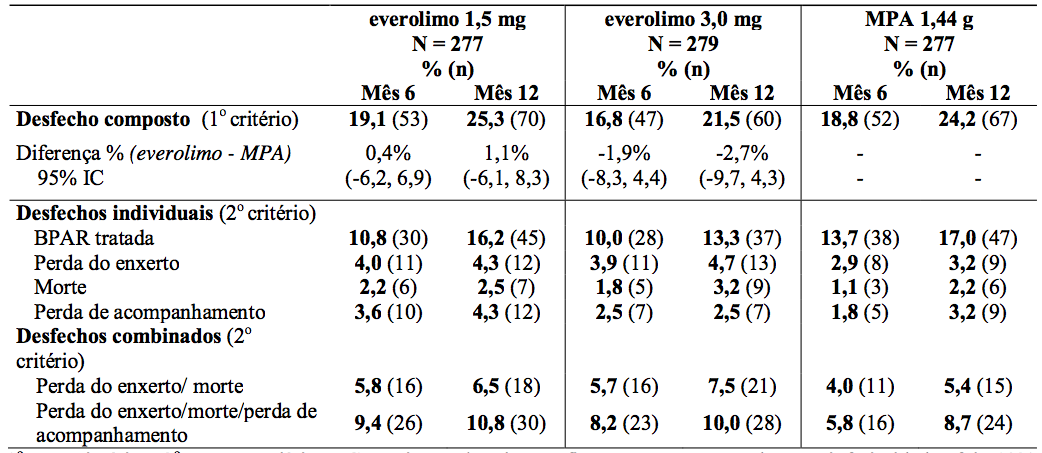
1° = primário, 2° = secundário, IC = intervalo de confiança, a margem de não-inferioridade foi 10%.
Desfecho composto: rejeição aguda comprovada por biópsia (BPAR), perda de enxerto, morte ou perda de acompanhamento.
Alterações na função renal, como demonstradas pelo cálculo da taxa de filtração glomerular (TFG), utilizando a fórmula MDRD, estão apresentadas na Tabela 4.
A proteinúria foi avaliada em visitas agendadas por meio de análise local de proteína urinária/creatinina e categorizadas por níveis de relevância clínica como representado na Tabela 5. Poucos pacientes em qualquer um dos grupos de tratamento alcançaram o limiar nefrótico, mas houve uma proporção consistentemente maior de pacientes que receberam Everolimo (substância ativa) na categoria subnefrótica que no caso do grupo MPA. Um efeito de concentração mostrou-se relacionando aos níveis de proteinúria para níveis mínimos de Everolimo (substância ativa) particularmente em valores de Cmín acima de 8 ng/mL. As reações medicamentosas adversas relatadas com regime de Everolimo (substância ativa) foram incluídas abaixo (Tabela 17). Foi relatada uma frequência menor de infecção viral nos pacientes tratados com Everolimo (substância ativa) resultante principalmente das menores taxas de notificação de infecção por CMV (0,7% versus 5,95%) e infecção pelo vírus BK (1,5% versus 4,8%).
Tabela 4: Estudo A2309: Função renal (TFG calculada usando MDRD) em 12 meses (população ITT):

Valor não inserido de TFG no mês 12: perda do enxerto = 0; morte ou perda do acompanhamento para função renal = LOCF1 (abordagem com base na observação mais recente 1: final do tratamento (até o mês 12)).
MDRD: Modificação da dieta na doença renal.
Tabela 5: Estudo A2309: Razão proteína urinária e creatinina:
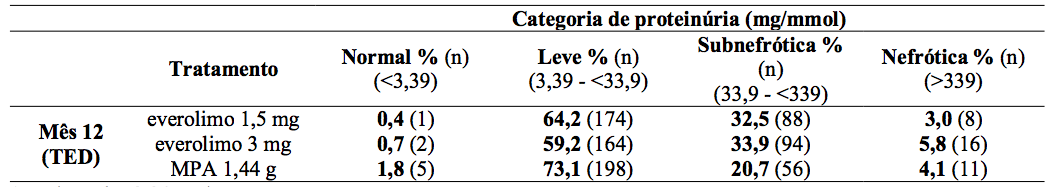
1mg/mmol = 8,84mg/g.
TED: Final do tratamento (valor do mês 12 ou abordagem com base na observação mais recente).
Transplante cardíaco
No estudo cardíaco de fase III (B253), foram estudados Everolimo (substância ativa) 1,5mg/dia e Everolimo (substância ativa) 3mg/dia em combinação com doses padrões de ciclosporina para microemulsão e corticosteroides versus azatioprina (AZA), 1-3mg/kg/dia. O desfecho primário foi composto por: incidência de rejeição aguda ? ISHLT grau 3A, rejeição aguda associada com comprometimento hemodinâmico, perda de enxerto, morte do paciente ou perda de acompanhamento nos meses 6, 12 e 24. A incidência de rejeição aguda confirmada por biópsia ? ISHLT grau 3A, no mês 6, foi de 27,8% para o grupo de 1,5mg/dia, 19% para o grupo de 3mg/dia e 41,6% para o grupo de AZA, respectivamente (p = 0,003 para 1,5mg versus controle, p <0,001 para 3mg versus controle).
Baseando-se em dados de ultrassom intravascular da artéria coronária obtidos de um subgrupo da população estudada, ambas as doses de Everolimo (substância ativa) foram estatística e significativamente mais eficazes do que AZA na prevenção da vasculopatia do aloenxerto (definida como um aumento na espessura máxima da íntima ?0,5 mm em relação ao valor basal, em pelo menos um corte de uma sequência automatizada), um importante fator de risco para perda do enxerto a longo prazo.
Nível sérico elevado de creatinina foi observado mais frequentemente em pacientes que estavam utilizando Everolimo (substância ativa) em combinação com doses plenas de ciclosporina para microemulsão do que em pacientes utilizando AZA. Este resultado indica que Everolimo (substância ativa) aumenta a nefrotoxicidade induzida pela ciclosporina. Entretanto, análises adicionais sugeriram que a função renal poderia ser melhorada com a redução da dose de ciclosporina sem perda da eficácia se os níveis sanguíneos de Everolimo (substância ativa) forem mantidos acima dos valores mínimos. Os estudos A2411 e A2310 foram posteriormente conduzidos para investigar isso.
O estudo A2411, aberto, randomizado, com duração de 12 meses, comparou Everolimo (substância ativa) em combinação com doses reduzidas de ciclosporina para microemulsão e corticosteroides ao micofenolato de mofetila (MMF) e doses padrão de ciclosporina para microemulsão e corticosteroides em pacientes receptores de transplante cardíaco “de novo”. O estudo incluiu um total de 174 pacientes. A dose inicial de Everolimo (substância ativa) (N = 92) foi de 1,5mg/dia e ajustada para manter o nível sanguíneo de Everolimo (substância ativa) entre 3-8 ng/mL. A MMF (N = 84) foi iniciada com uma dose de 1,500mg duas vezes ao dia.
As doses de ciclosporina para microemulsão foram ajustadas para atingir os seguintes níveis mínimos (ng/mL):
Tabela 6:
A função renal foi melhorada com a redução da dosagem de ciclosporina com clearance (depuração) de creatinina médio (fórmula de Cockcroft-Gault) em 6 meses: Everolimo (substância ativa): 65,4 v. MMF: 72,2 mL/mn, e em 12 meses: Everolimo (substância ativa): 68,7 v. MMF: 71,8 mL/mn. Eficácia, expressa como a taxa de episódios de rejeição aguda comprovados por biópsia (grau ISHLT ?3A), foi mantida como comparável nos dois grupos aos 12 meses (Everolimo (substância ativa): 22,8% v. MMF: 29,8%). O estudo A2310 é um estudo fase III, multicêntrico, aberto, randomizado que compara a eficácia e a segurança de dois tratamentos Everolimo (substância ativa)/ciclosporina dose reduzida, contra um tratamento padrão de micofenolato de mofetila (MMF)/ciclosporina por 24 meses. O uso da terapia de indução foi centro-específico, as opções sendo de não-indução ou indução com basiliximabe ou timoglobulina. Todos os pacientes receberam corticosteroides.
As doses iniciais nos dois grupos de Everolimo (substância ativa) foram de 1,5mg/dia e 3mg/dia, posteriormente modificadas a partir do dia 4 em diante, para manter os níveis sanguíneos mínimos de Everolimo (substância ativa) de 3 a 8 ng/mL e 6 a 12 ng/mL, respectivamente. A dose de MMF foi de 3 g/dia. As doses de ciclosporina foram adaptadas para manter o mesmo objetivo de intervalo dos níveis sanguíneos mínimos como no estudo A2411. As concentrações sanguíneas de ciclosporina e Everolimo (substância ativa) são apresentadas na Tabela 7.
O recrutamento para o tratamento experimental com Everolimo (substância ativa) no braço de dosagem superior foi interrompido prematuramente devido a um aumento da taxa de mortalidade dentro deste grupo de tratamento, causado por infecção e doenças cardiovasculares, que ocorreram dentro dos primeiros 90 dias pós-randomização. A natureza e o padrão das fatalidades neste braço de dosagem não sugerem que a diferença esteja vinculada à presença ou ao tipo de terapia de indução.
Comparações estatísticas são limitadas a comparações entre os braços do tratamento concluído. Os níveis de concentração sanguínea do medicamento efetivamente alcançados estão descritos na Tabela 7.
Tabela 7: Estudo A2310: Medição dos níveis mínimos sanguíneos de ciclosporina (CsA) e Everolimo (substância ativa):


Os números são médias ± DP dos valores medidos com C0 = nível mínimo.
O desfecho primário de eficácia foi uma variável composta de falha, implicando na ocorrência de qualquer das seguintes características: episódio de rejeição aguda comprovada por biópsia de ISHLT ? grau 3A, episódio de rejeição aguda (AR) associada com comprometimento hemodinâmico (HDC), perda de enxerto/retransplante, morte ou perda de acompanhamento. O resultado da eficácia em 12 meses é mostrado na Tabela 8.
Tabela 8: Estudo A2310: Taxas de incidência de desfecho de eficácia, por grupo de tratamento (População ITT - 12 meses de análise):
|
Desfechos de eficácia |
Everolimo (substância ativa) 1,5mg |
MMF |
|
Primário: falha de eficácia composta
|
99 (35,1) |
91 (33,6) |
|
Secundário: Perda de acompanhamento** |
33 (11,7) 11 (3,9) |
24 (8,9) 11 (4,1) |
Falha de eficácia composta: rejeição aguda comprovada por biópsia (BPAR) episódios de ISHLT grau ?3A, rejeição aguda (AR) associada com Comprometimento Hemodinâmico (HDC), perda do enxerto/retransplante, morte ou perda de acompanhamento.
* Perda de acompanhamento para desfechos relevantes (primário ou secundário).
A alta taxa de fatalidade no braço Everolimo (substância ativa) em relação ao braço MMF foi, principalmente, o resultado de aumento na taxa de fatalidades por infecção, nos três primeiros meses, entre os pacientes de Everolimo (substância ativa) no subgrupo do estudo de pacientes que receberam terapia de indução com timoglobulina. A incidência de 3 meses notavelmente alta de infecções graves em pacientes que receberam Everolimo (substância ativa) do que nos que receberam MMF no subgrupo com timoglobulina parece refletir maior potência imunossupressora. O desequilíbrio das fatalidades no subgrupo com timoglobulina, sendo particularmente evidente entre os pacientes internados antes do transplante e com dispositivos de assistência ventricular-L, sugere maior vulnerabilidade desses pacientes às consequências de complicações infecciosas.
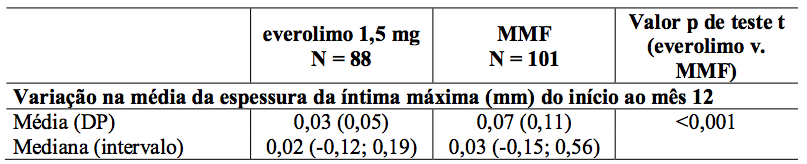
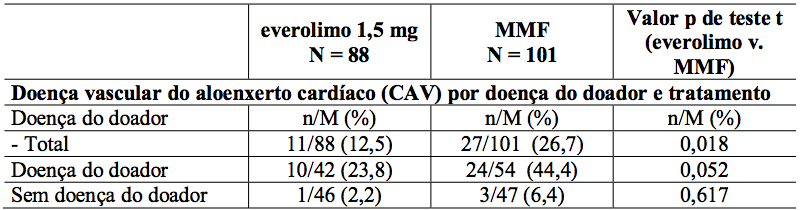
Estudos de ultrassonografia intravascular (IVUS) foram realizados em um subgrupo de pacientes para investigar mudanças pós-transplante (valor relativo do mês 12 a um valor basal efetivo durante os três primeiros meses pós-transplante) na espessura da íntima dentro de um segmento descendente anterior esquerdo (LAD) da artéria coronária. Os resultados da medida da variação da espessura da íntima máxima, juntamente com frequência de pacientes com doença vascular do aloenxerto cardíaco (definido como um aumento na espessura da íntima máxima de 0,5 mm ou mais) são descritos na Tabela 9.
Tabela 9: Alteração média da espessura da íntima máxima (mm) desde o início até o Mês 12 e incidência de doença vascular do aloenxerto cardíaco (CAV) por doença e tratamento dos doadores e tratamento (População IVUS - 12 meses de análise):
Avaliação da IVUS foi realizada até o dia 105.
O valor p para a variação do início deve ser comparado com o nível de significância de dois lados 0,025.
n = número de pacientes com um evento de CAV no estado de doença do doador; M = o número total de pacientes dentro do estado de doença do doador.
O reduzido aumento na espessura da íntima coronariana em pacientes usando Everolimo (substância ativa) em relação aos pacientes de MMF foi aparente independentemente da idade, sexo, presença ou ausência de diabetes e nível máximo de colesterol sérico observado no Mês 12.
A função renal ao longo do estudo A2310, avaliada pela taxa de filtração glomerular (TFG), calculada utilizando a fórmula MDRD, indica uma diferença estatisticamente significativa de 5,5 mL/min/1,73 m2 (97,5% IC -10,9; -0,2) inferior para o grupo de Everolimo (substância ativa) 1,5mg no Mês 12.
Os dados sugerem que a diferença observada foi associada principalmente à exposição à ciclosporina. Essa diferença foi reduzida para 3,6 mL/min/1,73 m2 e não estatisticamente significativa (97,5% IC -8,9, 1,8) em centros onde os níveis médios de ciclosporina foram menores nos pacientes que recebem Everolimo (substância ativa) do que em pacientes randomizados para o braço de controle, como recomendado.
Adicionalmente, a diferença foi impulsionada principalmente por uma diferença desenvolvida durante o primeiro mês pós-transplante, quando os pacientes ainda estão em uma situação de instabilidade hemodinâmica, possivelmente, confundindo a análise da função renal. Posteriormente, a diminuição da TFG média do Mês 1 ao Mês 12 foi significativamente menor no grupo Everolimo (substância ativa) do que no grupo controle (-6,4 vs -13,7 mL/min, p = 0,002).
Proteinúria, expressa como proteína urinária: níveis de creatinina urinária medidos em amostras de urina tenderam ser mais elevada nos pacientes tratados com Everolimo (substância ativa). Os valores subnefróticos foram observados em 22% dos pacientes que receberam Everolimo (substância ativa) comparados aos pacientes MMF (8,6%); níveis nefróticos também foram relatados (0,8%), representando 2 pacientes em cada grupo de tratamento.
As reações adversas para o grupo de 1,5mg de Everolimo (substância ativa) no estudo A2310 são consistentes com reações adversas a medicamentos apresentados na Tabela 17. Uma menor taxa de infecções virais foi relatada em pacientes tratados com Everolimo (substância ativa) resultando principalmente em uma menor taxa de relatos de infecção pelo CMV em comparação com MMF (7,2% versus 19,4%).
Transplante hepático
Em um estudo fase III (H2304) de transplante hepático em adultos, exposição reduzida à tacrolimo e Everolimo (substância ativa) 1,0mg duas vezes ao dia foram administrados a pacientes HCV+ e HCV-, com dose inicial de Everolimo (substância ativa) administrada aproximadamente 4 semanas após o transplante e investigada versus exposição padrão ao tacrolimo. A dose de Everolimo (substância ativa) foi ajustado para manter níveis sanguíneos mínimos de Everolimo (substância ativa) entre 3-8 ng/mL para o braço de Everolimo (substância ativa) + tacrolimo reduzido. Os níveis mínimos médios de Everolimo (substância ativa) estiveram dentro da variação alvo durante todo o tempo, variando entre 3,4 a 6,3 ng/mL no braço de Everolimo (substância ativa) + tacrolimo reduzido. As doses de tacrolimo foram subsequencialmente ajustadas para atingir os níveis mínimos alvo entre 3 a 5 ng/mL por 12 meses no braço de Everolimo (substância ativa) + tacrolimo reduzido.
O desfecho primário do estudo foi comparar a taxa de eficácia/falha, definido como o desfecho composto de rejeição aguda comprovada por biópsia tratada, perda do enxerto ou morte com minimização prematura de tacrolimo, facilitada por introdução de Everolimo (substância ativa) iniciado aproximadamente 4 semanas após o transplante hepático, até a exposição padrão à tacrolimo no mês 12.
No geral, durante os 12 meses de análise, a incidência do desfecho composto (BPAR tratada, perda de enxerto ou morte) foi menor no braço de Everolimo (substância ativa) + tacrolimo reduzido (6,7%) comparado ao braço controle de tacrolimo (9,7%) (Tabela 10). A diferença nas estimativas entre Everolimo (substância ativa) + tacrolimo reduzido e tacrolimo controle foi de -3,0%, com IC 97,5%: (-8,7% a 2,6%). Em relação às taxas de perda de enxerto e casos fatais, o braço de Everolimo (substância ativa) + tacrolimo reduzido foi não-inferior se comparado ao braço tacrolimo controle, indicando risco de mortalidade não aumentado nesta população. Uma taxa significativa e estatisticamente menor de rejeição aguda foi observada no braço de Everolimo (substância ativa) + tacrolimo reduzido (3,7%), se comparado ao braço controle (10,7%) (Tabela 11). Os resultados são similares entre pacientes HCV+ e HCV-.
Tabela 10: Estudo H2304: Comparação entre os grupos de tratamento para as taxas de incidência Kaplan-Meier (KM) de desfechos primários de eficácia (população ITT – 12 meses de análise):
|
Estatística |
EVR+TAC reduzido |
Controle TAC |
|
Número de eficácia/falha composta (tBPAR, perda de | 16 |
23 |
|
KM estimado da taxa de incidência de eficácia/falha composta (tBPAR, perda de enxerto ou morte) no Mês 12 | 6,7% | 9,7% |
|
Diferença nas estimativas de KM (versus controle) |
-3,0% | |
|
IC 97,5% para diferença |
(-8,7%; 2,6%) | |
|
Valor P do teste Z para (TAC reduzido - Controle = 0) (teste de não-diferença) |
0,230 | |
|
Valor P* do teste Z para (TAC reduzido - Controle ? 0,12) (teste de não-inferioridade) |
<0,001 |
1. tBPAR = rejeição aguda comprovada por biópsia tratada. Os resultados laboratoriais locais de biópsia são usados para definir tBPAR.
2. *Valor P do teste Z para o teste de não-inferioridade (margem de não-inferioridade = 12%) é para um teste unilateral e foi comparado a um nível de significância de 0,0125.
3. Na estimativa de Kaplan-Meier, o dia censura para pacientes sem o evento é o dia de último contato.
Tabela 11: Estudo H2304: Comparação entre grupos de tratamento para taxas de incidência de desfechos de eficácia secundários (população ITT – 12 meses de análise):
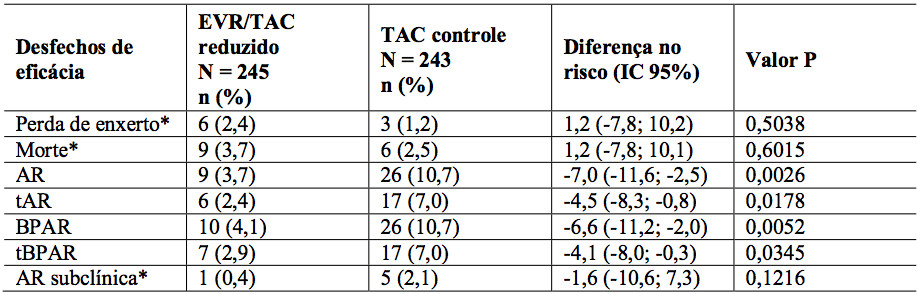
1. AR = Rejeição aguda; BPAR = rejeição aguda comprovada por biópsia; tBPAR = rejeição aguda comprovada por biópsia tratada. Os resultados laboratoriais locais de biópsia são usados para definir BPAR e tBPAR.
2. Perda de seguimento por “perda de enxerto, morte ou perda de seguimento” é definida como um paciente que não morreu, não apresentou perda de enxerto e cujo último dia de contato é anterior ao limite inferior da janela de visita do mês 12.
3. * = intervalo de confiança exato e teste de Fisher bilateral exato utilizados para essas variáveis. Para outras, intervalo de confiança assintomático e teste Chi-quadrado de Pearson são usados.
4. Todos os valores P são para teste bilateral e foram comparados a um nível de significância de 0,05.
A comparação entre os grupos de tratamento para mudança no eGFR (MDRD4) [mL/min/1,73 m2] do tempo de randomização (dia 30) ao mês 12 para a população ITT está representada na Tabela 12. A diferença média ajustada entre o braço de Everolimo (substância ativa) + tacrolimo reduzido e braço tacrolimo controle no eGFR ao mês 12 foi de 8,50 mL/min/1,73 m2 (P <0,001; IC 97,5%: 3,74; 13,27). Um eGFR aumentado foi observado durante o estudo e no mês 12 para EVR + TAC reduzido (80,9 mL/min/1,73 m2) em comparação ao TAC controle (70,3 mL/min/1,73 m2).
Tabela 12: Estudo H2304: Comparação entre grupos de tratamento para eGFR (MDRD4) no mês 12 (população ITT – 12 meses de análise):
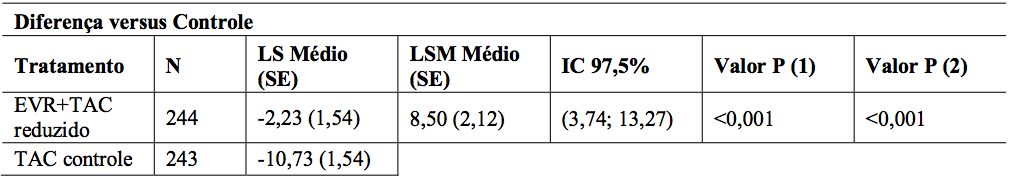
1. Mínimos quadrados significam intervalos de confiança de 97,5%, valores P são de um modelo ANCOVA contendo tratamento e estado HCV como fatores, e eGFR inicial como covariável.
2. Regras de imputação de eGFR ausente no mês 12 (MDRD4) valores:
- Usam o último valor disponível antes da randomização para pacientes com não eGFR pós-randomização;
- Usam o valor mínimo se o último valor é observado entre a randomização e o mês 6;
- Ou usam o mínimo valor entre mês 6 e mês 12 se o último valor é observado no mês 6 ou após;
- Usam 15 mL/min/1,73 m2 se o paciente estava em diálise após a randomização.
3. Valor P (1): Teste de não-inferioridade com margem NI = -6 mL/min/1,73 m2, com nível unilateral de 0,0125. 4. Valor P (2): Teste de superioridade com níveis bilaterais de 0,025.
Características Farmacológicas
Farmacodinâmica
Grupo farmacoterapêutico: agente imunossupressor seletivo.
Código ATC: L04A A18.
Mecanismo de ação
O Everolimo (substância ativa) é um inibidor do sinal de proliferação que previne a rejeição ao aloenxerto em modelos de alotransplantes em roedores e primatas não humanos. Exerce efeito imunossupressor pela inibição da proliferação das células T ativadas por antígenos e, consequentemente, da expansão clonal, controladas por interleucinas específicas de células T como interleucina-2 e interleucina-15. O Everolimo (substância ativa) inibe uma via de sinalização intracelular que normalmente leva à proliferação celular quando desencadeada pela ligação desses fatores de crescimento de células T aos seus respectivos receptores. O bloqueio deste sinal pelo Everolimo (substância ativa) faz com que as células estacionem no estágio G1 do ciclo celular.
No nível molecular, o Everolimo (substância ativa) forma um complexo com a proteína citoplasmática FKBP-12. Na presença do Everolimo (substância ativa), a fosforilação estimulada pelo fator de crescimento da p70 S6 quinase é inibida. Uma vez que a fosforilação da p70 S6 quinase está sob controle da FRAP (também chamada de m-TOR), esta descoberta sugere que o complexo de Everolimo (substância ativa)-FKBP-12 se liga e, assim, interfere com a função da FRAP.
A FRAP é uma proteína regulatória chave que controla o metabolismo, crescimento e proliferação celular; o bloqueio da função da FRAP explica a interrupção do ciclo celular causada pelo Everolimo (substância ativa).
Portanto, o Everolimo (substância ativa) tem um mecanismo de ação diferente da ciclosporina. Em modelos pré-clínicos de alotransplantes, a combinação de Everolimo (substância ativa) e ciclosporina foi mais eficaz do que cada fármaco sozinho.
O efeito do Everolimo (substância ativa) não se restringe às células T. O Everolimo (substância ativa) geralmente inibe a proliferação de células hematopoiéticas estimulada por fatores de crescimento e de células não hematopoiéticas, tais como as células de músculo liso vascular. A proliferação de células de músculo liso vascular estimulada por fatores de crescimento, que é induzida por lesão das células endoteliais e que leva à formação da neointima, desempenha um papel fundamental na patogênese da rejeição crônica. Estudos pré-clínicos com Everolimo (substância ativa) mostraram a inibição da formação da neointima em um modelo de alotransplante de aorta em ratos.
Farmacocinética
Absorção
O pico da concentração de Everolimo (substância ativa) ocorre 1 a 2 horas após administração de uma dose oral. As concentrações sanguíneas do Everolimo (substância ativa) em pacientes receptores de transplante são proporcionais à dose, sobre o intervalo de dose de 0,25mg a 15mg. Efeito da alimentação: quando a forma farmacêutica comprimidos é administrada com uma refeição rica em lipídeos, a Cmáx e a ASC de Everolimo (substância ativa) são reduzidas em 60% e 16%, respectivamente. Para minimizar a variabilidade, Everolimo (substância ativa) deve ser administrado sempre da mesma maneira com ou sem alimentos.
Distribuição
A proporção sangue-plasma de Everolimo (substância ativa), que é dependente da concentração dentro da faixa de 5 a 5.000 ng/mL, varia de 17% a 73%. A ligação às proteínas plasmáticas é de aproximadamente 74% em voluntários sadios e pacientes com insuficiência hepática moderada. O volume de distribuição associado com a fase final (Vz/F) em pacientes receptores de transplante renal em manutenção é de 342 ± 107 L.
Biotransformação/metabolismo
O Everolimo (substância ativa) é um substrato do CYP3A4 e glicoproteína-P. Após a administração oral, este é o principal componente circulante no sangue humano. Seis metabólitos principais de Everolimo (substância ativa) foram detectados no sangue humano, incluindo três metabólitos mono-hidroxilados, dois produtos hidrolíticos de anel aberto e um conjugado fosfatidilcolina de Everolimo (substância ativa). Estes metabólitos foram também identificados em várias espécies de animais utilizadas nos estudos de toxicidade e mostrou aproximadamente 100 vezes menos atividade do que o próprio Everolimo (substância ativa). Assim, a substância mãe contribui para a maior parte da atividade farmacológica total do Everolimo (substância ativa).
Excreção
Após a administração de dose única de Everolimo (substância ativa) radiomarcado a pacientes receptores de transplante recebendo ciclosporina, a maioria da radioatividade (80%) foi recuperada nas fezes e apenas uma quantidade menor (5%) foi excretada na urina. Não foi detectada substância mãe na urina ou nas fezes.
Farmacocinética no estado de equilíbrio
A farmacocinética foi comparável em pacientes receptores de transplante renal e cardíaco recebendo Everolimo (substância ativa) duas vezes ao dia simultaneamente com ciclosporina para microemulsão. O estado de equilíbrio é alcançado no 4o dia com acúmulo nos níveis sanguíneos de 2 a 3 vezes comparado com a exposição após a primeira dose. O tmáx ocorre de 1 a 2 horas após a administração da dose. Com 0,75mg e 1,5mg duas vezes ao dia, as Cmáx médias observadas foram de 11,1 ± 4,6 e 20,3 ± 8,0 ng/mL, respectivamente, e as ASC médias foram de 75 ± 31 e 131 ± 59 ng.h/mL, respectivamente. Com 0,75mg e 1,5mg duas vezes ao dia, as concentrações médias sanguíneas mínimas pré-dose (Cmín) foram 4,1 ± 2,1 e 7,1 ± 4,6 ng/mL, respectivamente.
A exposição ao Everolimo (substância ativa) permanece estável com o tempo no primeiro ano após o transplante. A Cmín está significativamente correlacionada com a ASC produzindo um coeficiente de correlação entre 0,86 e 0,94. Baseado na análise farmacocinética da população, o clearance (depuração) oral (CL/F) é de 8,8 L/h (27% de variação interpaciente) e o volume de distribuição central (Vc/F) é 110 L (36% de variação interpaciente). A variabilidade residual na concentração sanguínea é 31%. A meia-vida de eliminação é de 28 ± 7 h.
Insuficiência hepática
Em relação à ASC de Everolimo (substância ativa) em indivíduos com função hepática normal, a ASC média de 6 pacientes com insuficiência hepática leve (Child-Pugh Classe A) foi 1,6 vezes maior; em dois grupos estudados de forma independente com 8 e 9 pacientes com insuficiência hepática moderada (Child-Pugh Classe B), a ASC média foi 2,1 vezes e 3,3 vezes maior; e em 6 pacientes com insuficiência hepática grave (Child-Pugh Classe C), a ASC média foi 3,6 vezes maior.
Significa que as meias-vidas foram 52, 59 e 78 horas na insuficiência hepática leve, moderada e grave. As meias-vidas prolongadas atrasam o tempo para atingir o estado de equilíbrio nos níveis sanguíneos de Everolimo (substância ativa).
Insuficiência renal
Insuficiência renal pós-transplante (faixa de clearance (depuração) de creatinina, 11 – 107 mL/min) não afetou a farmacocinética do Everolimo (substância ativa).
Pediatria
O CL/F do Everolimo (substância ativa) aumentou de modo linear com a idade do paciente (1 a 16 anos), superfície de área corpórea (0,49-1,92 m2) e peso (11-77 kg) dos pacientes. O estado de equilíbrio CL/F foi de 10,2 ± 3,0 L/h/m2 e a meia-vida de eliminação foi de 30 ± 11 h.
Idosos
Uma redução limitada do clearance (depuração) oral do Everolimo (substância ativa) de 0,33% ao ano foi estimada em adultos (faixa de idade estudada foi 16 a 70 anos). O ajuste de dose não é considerado necessário.
Etnia
Baseado na análise farmacocinética da população, o clearance (depuração) oral (CL/F) é, em média, 20% maior em pacientes negros receptores de transplante.
Relação exposição-resposta
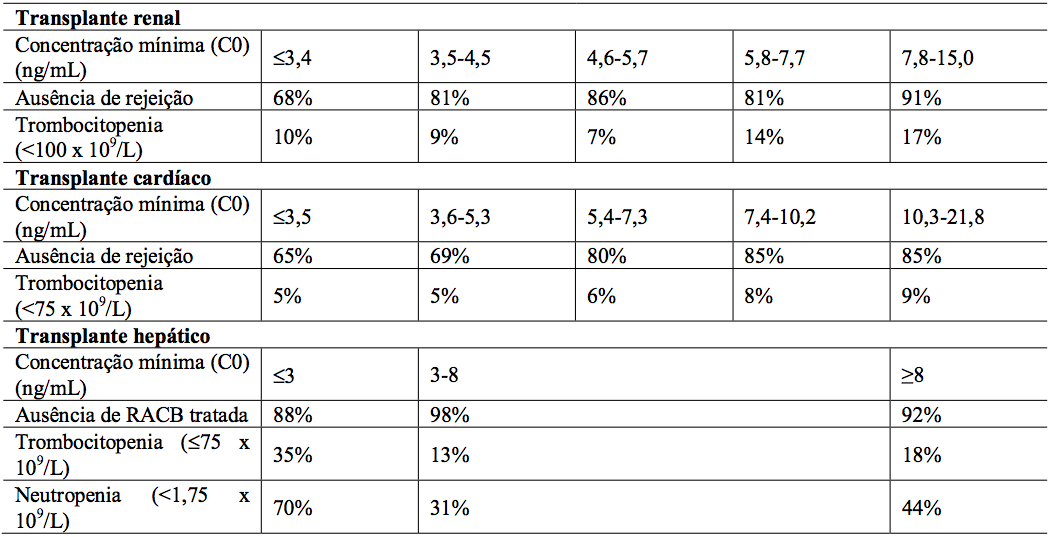
A média da concentração mínima de Everolimo (substância ativa) nos 6 primeiros meses pós-transplante foi relacionada à incidência de rejeição aguda confirmada por biópsia e à trombocitopenia em pacientes receptores de transplante renal e cardíaco (vide Tabela 13). Em pacientes com transplante hepático, a relação dos níveis mínimos de Everolimo (substância ativa) e eventos clínicos não é bem estabelecida, entretanto, altas exposições não estão relacionadas com aumento de eventos adversos.
Tabela 13: Relação exposição-resposta para Everolimo (substância ativa) em pacientes transplantados:
Dados de segurança pré-clínicos
O perfil de segurança pré-clínico do Everolimo (substância ativa) foi avaliado em camundongos, ratos, mini-porcos, macacos e coelhos.
Os principais órgãos-alvo foram os sistemas reprodutores femininos e masculinos (degeneração tubular testicular, contagem reduzida de esperma no epidídimo e atrofia uterina) em diversas espécies e, somente em ratos, os pulmões (aumento de macrófagos alveolares) e olhos (opacidade da linha da sutura lenticular anterior). Alterações menores no rim foram observadas em ratos (exacerbação de lipofuscina idade-dependente no epitélio tubular) e em camundongos (exacerbação de lesões secundárias). Não há indicações de toxicidade renal em macacos ou mini porcos.
O Everolimo (substância ativa) parece exacerbar a ocorrência espontânea de outras doenças de base (miocardite crônica em ratos, infecção por vírus coxsackie no plasma e coração em macacos, infestação coccidial do trato gastrintestinal em mini-porcos, lesões na pele em camundongos e macacos). Estes fatos foram observados pela exposição sistêmica em níveis dentro da faixa terapêutica de exposição ou acima, com exceção das ocorrências em ratos que ocorreram abaixo da exposição terapêutica devido à alta distribuição tecidual.
A ciclosporina em combinação com Everolimo (substância ativa) causou maior exposição sistêmica ao Everolimo (substância ativa) e aumentou a toxicidade. Não houve órgão-alvo novo em ratos. Macacos apresentaram hemorragia e arterite em muitos órgãos.
Em um estudo de fertilidade em ratos machos, a morfologia testicular foi afetada com ?0,5mg/kg; e a motilidade dos espermatozoides, contagem de cabeças de espermatozoides e níveis de testosterona plasmática foram diminuídos com 5mg/kg, que está dentro da faixa de exposição terapêutica e causou uma diminuição da fertilidade nos machos. Houve evidência de reversibilidade. A fertilidade das fêmeas não foi afetada, porém o Everolimo (substância ativa) cruzou a placenta e foi tóxico para o embrião. Em ratos, o Everolimo (substância ativa) causou embrio/fetotoxicidade, com exposição sistêmica abaixo da faixa terapêutica, que foi manifestada como mortalidade e peso fetal reduzido. A incidência de variações esqueléticas e malformações com 0,3 e 0,9mg/kg (por exemplo, fissura no esterno) foi aumentada. Em coelhos, a embriotoxicidade foi evidente pelo aumento na reabsorção tardia.
Estudos de genotoxicidade que levaram em consideração todos os desfechos relevantes não mostraram evidência de atividade clastogênica ou mutagênica. A administração de Everolimo (substância ativa) durante 2 anos para camundongos e ratos não indicou qualquer potencial oncogênico até as maiores doses correspondentes respectivamente a 8,6 e 0,3 vezes a exposição clínica estimada.
Cuidados de Armazenamento
Você deve guardar os comprimidos de Certican em temperatura ambiente (entre 15 e 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Certican é um comprimido redondo, branco a levemente amarelado.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Mensagens de Alerta
- Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
- Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS – 1.0068.0959
Farm. Resp.: Flavia Regina Pegorer - CRF-SP 18.150
Importado por:
Novartis Biociências S.A.
Av. Prof. Vicente Rao, 90
São Paulo - SP
CNPJ: 56.994.502/0001-30
Indústria Brasileira
Fabricado por:
Novartis Pharma Stein AG, Stein, Suíça.
Venda sob prescrição médica.