para o que é indicado e para que serve?
Para que serve Você deve ler atentamente esta bula antes de começar o tratamento com Eprex.Continue lendo...
ofertas de
Eprex - Alfaepoetina 4000...
ofertas de Eprex - Alfaepoetina 4000...
R$ 460,00
R$ 1.210,64
R$ 1.670,00
R$ 1.736,80
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Você deve ler atentamente esta bula antes de começar o tratamento com Eprex. Estas informações não substituem as orientações do seu médico.
Você e seu médico devem conversar a respeito das suas condições médicas antes do início e durante o tratamento com Eprex.
Se houver alguma informação que você não entenda ou se você necessitar de esclarecimentos adicionais, consulte o seu médico, que ele poderá orientá-lo.
Eprex está indicado
- No tratamento da anemia secundária a insuficiência renal crônica, em pacientes pediátricos e adultos em diálise ou em fase pré-diálise;
- No tratamento da anemia associada ao câncer não mieloide e secundária a quimioterapia mielossupressora;
- Na anemia do paciente com AIDS e submetido ao tratamento com zidovudina (AZT);
- No programa de doação sanguínea autóloga para facilitar a coleta de sangue autólogo e diminuir o risco de transfusões alogênicas em pacientes com anemia moderada (hemoglobina entre 10-13 g/dL e sem deficiência de ferro). Esses pacientes serão submetidos à cirurgia eletiva de grande porte onde se estima uma necessidade transfusional elevada (mais de 4 unidades para o sexo feminino e mais de 5 unidades para o sexo masculino);
- Para aumentar os níveis de hemoglobina no período pré-operatório, evitando-se transfusões autólogas, em pacientes adultos que serão submetidos a cirurgias ortopédicas de grande porte. A anemia deve ser moderada (hemoglobina entre 10-13 g/dL), o paciente não deve estar em programa de doação sanguínea autóloga e a perda de sangue esperada deve ser moderada (900-1800 mL).
Como Exprex funciona?
A eficácia de Eprex é medida pelo aumento da hemoglobina (quantidade de células vermelhas no sangue).
O aumento da hemoglobina não é imediato. Geralmente leva algumas semanas para que a hemoglobina comece a aumentar.
O tempo e a dose de Eprex necessários para promover o aumento variam de acordo com cada paciente.
Contraindicação
Os pacientes que desenvolveram anticorpos em virtude de Aplasia Pura de Células Vermelhas por tratamento com qualquer eritropoetina, não devem receber Eprex ou qualquer outra eritropoetina.
Hipersensibilidade (alergia) a qualquer componente da fórmula do medicamento.
Todas as contraindicações associadas aos programas de pré-doação de sangue autólogo devem ser respeitadas em pacientes recebendo Eprex.
Pressão alta não controlada com medicamento.
Doença grave no coração, nas artérias, nas carótidas ou doença vascular cerebral, incluindo infarto do miocárdio ou acidente vascular cerebral recentes, em pacientes que serão submetidos à cirurgia ortopédica de grande porte e não serão incluídos em programa de doação sanguínea autóloga.
Qualquer razão em que o paciente cirúrgico não possa receber profilaxia adequada com antitrombóticos.
Como usar
xRecomenda-se que a aplicação seja feita por uma pessoa treinada por um profissional de saúde.
O medicamento é para apenas uma única aplicação. O medicamento não deve ser usado e deve ser descartado se o lacre estiver rompido, o líquido apresentar coloração ou partículas em suspensão, o medicamento possa ter sido congelado ou se houve falha na refrigeração.
Uso Subcutâneo
Uso intravenoso
A administração por esta via deve ser efetuada por profissionais de saúde.
A injeção deve ser aplicada durante 1 a 5 minutos, dependendo da dose total.
Em pacientes em hemodiálise, a medicação deve ser aplicada durante ou após a sessão de diálise.
Para lavar o sistema de administração e garantir uma injeção satisfatória do medicamento na circulação, a injeção deve ser seguida por 10 mL de solução salina.
Injeções mais lentas, durante 5 minutos, podem ser benéficas em pacientes que apresentem efeitos colaterais do tipo gripal.
Eprex não deve ser administrado em infusão ou combinado a outras soluções parenterais.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Posologia
Eprex pode ser administrado por via intravenosa ou subcutânea.
Pacientes com insuficiência renal crônica
Em pacientes com insuficiência renal crônica e acesso intravenoso disponível (pacientes em hemodiálise), a administração de Eprex por via intravenosa é preferível. Se o acesso intravenoso não estiver disponível (pacientes ainda não submetidos à diálise ou em diálise peritoneal), Eprex pode ser administrado por via subcutânea.
A concentração de hemoglobina ideal deve ser de 10 a 12 g/dL em adultos e de 9,5 a 11 g/dL em crianças.
Em pacientes com insuficiência renal crônica a concentração de manutenção da hemoglobina não deve exceder o limite superior da faixa de concentração da hemoglobina.
Quando se altera a via de administração, a mesma dose deve ser usada inicialmente, e, então, deve ser titulada para manter a hemoglobina na faixa de concentração da hemoglobina.
Na fase de correção da anemia, a dose de Eprex deve ser aumentada se a hemoglobina não aumentar pelo menos 1 g/dL/mês.
Um aumento clinicamente significativo na hemoglobina geralmente não é observado em menos de 2 semanas e pode requerer até 6 - 10 semanas em alguns pacientes.
Quando a concentração de hemoglobina estiver dentro da faixa, a dose deve ser diminuída em 25 UI/kg/dose, para evitar exceder a faixa de concentração da hemoglobina.
A dose deve ser reduzida quando a hemoglobina se aproximar de 12g/dL. Além disso, se a concentração de hemoglobina exceder 12 g/dL, a terapia deve ser descontinuada (interrompida).
Em pacientes com insuficiência renal crônica, a concentração de hemoglobina não deve exceder 12 g/dL. Níveis de hemoglobina superiores a 12 g/dL podem estar associados com maior risco de eventos cardiovasculares, incluindo morte.
Reduções da dose podem ser feitas através da omissão (não utilização) de uma das doses semanais ou pela redução da quantidade de cada dose.
Pacientes adultos em hemodiálise
Em pacientes em hemodiálise, com acesso intravenoso disponível a administração de Eprex por via intravenosa é preferível.
Pacientes adultos em diálise peritoneal
Em pacientes sob diálise peritoneal, sem acesso intravenoso disponível, Eprex pode ser administrado por via subcutânea.
Pacientes adultos em pré-diálise (Pacientes adultos em estágio final de insuficiência renal)
Em pacientes com insuficiência renal ainda não submetidos à diálise, sem acesso intravenoso disponível, Eprex pode ser administrado por via subcutânea.
Pacientes pediátricos em hemodiálise
Em estudos clínicos, as seguintes doses de manutenção foram observadas após 6 meses de tratamento:
| Peso (kg) | Mediana da dose | Dose (UI/kg administrada 3x / semana) Dose usual de manutenção |
| < 10 | 100 | 75-150 |
| 10-30 | 75 | 60-150 |
| > 30 | 33 | 30-100 |
Os dados disponíveis sugerem que os pacientes com valores basais de hemoglobina muito baixos (hemoglobina < 6,8 g/dL) podem requerer doses de manutenção maiores que os pacientes com hemoglobina inicial mais alta (hemoglobina > 6,8 g/dL).
Pacientes adultos com câncer
A via subcutânea deve ser usada.
A faixa de concentração da hemoglobina deve ser de 10 a 12 g/dL em homens e mulheres e não deve ser excedida.
O tratamento com Eprex deve ser mantido até um mês após o término da quimioterapia. Entretanto, a necessidade de continuar o tratamento deve ser reavaliada periodicamente.
A dose inicial para o tratamento da anemia deve ser de 150 UI/kg, 3 vezes por semana.
Alternativamente, Eprex pode ser administrado por via subcutânea em uma dose inicial de 40.000 IU uma vez por semanda.
Se após 4 semanas de tratameno com a dose inicial, o nível de hemoglobina aumentou em pelo menos 1g/dL, ou a contagem de reticulócitos aumentou acima do basal em ? 40.000 IU células/?L, a dose deve permanecer a mesma.
Se após 4 semanas de tratamento com a dose inicial, a hemoglobina não aumentou em ? 1g/dL, e a contagem de reticulócitos não aumentou acima do basal em ? 40.000 células/?L, na ausência de transfusão sanguínea de células vermelhas, a dose deve ser aumentada para 300 UI/kg 3 vezes por semana ou 60.000 UI por semana.
Se após 4 semanas de tratamento de terapia adicional com 300 UI/kg 3 vezes por semana ou 60.000 UI por semana, a hemoglobina aumentou ? 1g/dL ou a contagem de reticulócitos aumentou ? 40.000 células/?L, a dose deve permanecer a mesma.
Se, após 4 semanas de terapia com 300 UI/kg 3 vezes por semana, a hemoglobina tiver aumentado menos que 1 g/dL, a resposta do indivíduo ao Eprex é improvável e o tratamento deve ser descontinuado.
Uma taxa de aumento de hemoglobina > 1 g/dL por 2 semanas ou 2 g/dL por mês ou níveis de hemoglobina > 12 g/dL devem ser evitados. Se os níveis de hemoglobina são aumentados em mais de 1 g/dL por 2 semanas ou 2 g/dL por mês ou se a hemoglobina estiver próxima de 12 g/dL, deve-se reduzir a dose de Eprex em cerca de 25 - 50% dependendo da taxa de aumento da hemoglobina.
Se a hemoglobina exceder 12 g/dL, a terapia deve ser descontinuada até que estes níveis caiam para 12 g/dL e então, reinstitue-se a terapia com Eprex com uma dose 25% abaixo da dose prévia.
Pacientes portadores de AIDS tratados com zidovudina (AZT)
Antes do início do tratamento com Eprex, recomenda-se que o nível de eritropoetina sérica seja determinado antes da transfusão. Os dados disponíveis sugerem que os pacientes com níveis séricos de eritropoetina > 500 mUI/mL provavelmente não responderão à terapia com Eprex.
Programa de doação de sangue autólogo em pacientes adultos a serem submetidos à cirurgia
Eprex deve ser administrado após o término de cada procedimento de doação, por via endovenosa.
Para pacientes que necessitam um menor grau de estimulação da eritropoese, um regime de 150-300 UI/g, duas vezes por semana, demonstrou aumentar a pré-doação autóloga e diminuir o declínio subsequente no hematócrito.
Para pacientes com anemia leve (hemoglobina entre 10-13 g/dL) que necessitam de pré-depósito de pelo menos 4 unidades de sangue, recomenda-se a posologia de 600 UI/kg por intravenosa, duas vezes por semana, por 3 semanas antes da cirurgia.
Pacientes em pré-operatório (que não participam de programa de doação de sangue autólogo)
Deve ser usada a via subcutânea de administração.
A dose recomendada é de 600 UI/kg de Eprex, por semana, durante três semanas antes da cirurgia (dias -21, -14 e -7) e no dia da cirurgia. Caso a cirurgia tenha indicação médica de ocorrer em menos de 3 semanas, a dose de 300 UI/kg deve ser administrada diariamente durante dez dias consecutivos, antes da cirurgia, no dia da cirurgia e nos quatro dias imediatamente posteriores à mesma.
Esta dose é recomendada para níveis de hemoglobina <13 g/dL.
A administração deve ser interrompida caso a hemoglobina atinja 15 g/dL ou acima e doses adicionais não devem ser administradas.
Populações especiais
Tratamento de pacientes pediátricos infectados pelo HIV tratados com zidovudina
A segurança e eficácia de Eprex em pacientes pediátricos infectados pelo HIV tratados com zidovudina não foram estabelecidas.
Tratamento de pacientes pediátricos a serem submetidos à cirurgia em um programa de pré-doação autóloga
A segurança e eficácia de Eprex em pacientes pediátricos a serem submetidos a cirurgia em um programa de pré-doação autóloga não foram estabelecidas.
Tratamento de pacientes pediátricos a serem submetidos à cirurgia ortopédica eletiva de grande porte
A segurança e eficácia de Eprex em pacientes pediátricos em programação de cirurgia programados para cirurgia ortopédica eletiva de grande porte não foram estabelecidas.
Idosos (65 anos de idade ou mais)
A seleção e o ajuste de dose para pacientes idosos devem ser individualizados para atingir e manter a faixa de concentração da hemoglobina.
O que fazer quando eu me esquecer de usar Eprex?
Faça a próxima injeção assim que você se lembrar. Se estiver faltando apenas um dia para a sua próxima injeção, pule a dose esquecida e continue com a sua programação normal de aplicação. Não dobre o número de injeções.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Eprex não deve ser usado:
- Após o prazo de validade do medicamento;
- Se o lacre estiver rompido;
- Se o líquido apresentar coloração ou partículas em suspensão;
- Se você souber ou achar que Eprex pode ter sido acidentalmente congelado;
- Se houver uma falha no funcionamento da geladeira;
- Se você souber ou suspeitar que Eprex tenha sido deixado a temperatura ambiente por mais de 60 minutos antes da injeção.
Advertências
Avise ao seu médico se você apresentar ou já tiver apresentado alguma das seguintes condições:
- Hipertensão arterial (pressão arterial aumentada);
- Doença do coração (como angina);
- Distúrbios da circulação sanguínea, resultando em pontadas e agulhadas, mãos ou pés frios ou câimbras musculares nas pernas;
- Distúrbios de coagulação sanguínea;
- Ataques epilépticos ou convulsões;
- Câncer;
- Anemia de outras causas;
- Insuficiência hepática.
Informe ao seu médico se você não puder utilizar medicamentos para “afinar” o sangue.
Se você for atendido em um hospital ou por outro médico para qualquer tratamento ou for submetido a um exame de sangue, lembre-se de informar que você está usando Eprex, pois Eprex pode alterar os resultados.
Informe ao seu médico se você apresenta quaisquer outros problemas médicos, uma vez que eles podem interferir no uso de Eprex.
Mulheres com insuficiência renal crônica podem ter a menstruação interrompida. Algumas destas mulheres podem ter reinício do ciclo menstrual após correção da anemia com Eprex. Assim, antes de iniciar o uso de Eprex, as mulheres devem conversar com seu médico sobre a necessidade de usar métodos para prevenir a gravidez.
Lembre-se de informar ao seu médico se você usou Eprex ou outro medicamento a base de epoetina no passado e se sua anemia piorou.
Informe ao seu médico imediatamente se você começar a sentir cansaço, tontura ou falta de ar. Estes sintomas podem significar resposta inadequada à alfaepoetina e as causas mais frequentes são deficiência de ferro, infecções, inflamações, deficiência de vitaminas, perda de sangue e outros.
Em pacientes com insuficiência renal, os sintomas de cansaço, tontura ou falta de ar podem raramente estar relacionados a baixa produção de células vermelhas pela medula óssea.
Apenas o seu médico saberá identificar o problema e resolvê-lo.
A pressão arterial deve ser adequadamente monitorada e controlada, se necessário, se você estiver recebendo alfaepoetina e o seu uso deve ser realizado com cautela na presença de hipertensão arterial não tratada, tratada de forma inadequada ou mal controlada.
Durante o tratamento com Eprex, pode ser necessário iniciar tratamento anti-hipertensivo ou aumentar a dose do anti-hipertensivo em uso. Caso a pressão arterial não seja controlada, interromper o uso do Eprex.
Crises hipertensivas com encefalopatia e convulsões que exigem a atenção imediata de um médico e cuidados médicos intensivos, também ocorreram durante o tratamento com Eprex em pacientes com pressão arterial prévia normal ou baixa.
Atenção especial deve ser dada ao aparecimento súbito de cefaleias tipo enxaqueca lancinante como um possível sinal de cuidado.
Eprex deve ser usado com cautela se você apresentar epilepsia, história de convulsões, ou condições médicas associadas a uma predisposição para atividade convulsiva, tais como infecções do SNC e metástases cerebrais.
Eprex deve ser usado com cautela se você apresentar insuficiência hepática crônica. A segurança da alfaepoetina não foi estabelecida em pacientes com disfunção hepática. Devido ao reduzido metabolismo, se você apresentar eritropoese (produção de eritrócitos) aumentada com a alfaepoetina.
A incidência aumentada de eventos trombóticos vasculares foi observada em pacientes recebendo agentes estimulantes de eritropoese (ESAs), incluindo tromboses arteriais e venosas e embolia (incluindo alguns desfechos fatais), tais como trombose venosa profunda, embolia pulmonar, trombose da retina e infarto do miocárdio. Adicionalmente, acidente vascular cerebral (incluindo infarto cerebral, hemorragia cerebral e ataques de isquemia transitória) foram relatados.
O risco relatado de eventos trombóticos vasculares deve ser avaliado contra os benefícios proporcionados pelo tratamento com Eprex, particularmente se você apresentar fatores de risco pré-existentes.
A sua concentração de hemoglobina deve ser monitorada com cautela devido ao potencial de risco aumentado de eventos tromboembólicos e casos fatais quando os pacientes são tratados em concentrações de hemoglobina acima da faixa para a indicação de uso.
A segurança e a eficácia do tratamento com Eprex não foram estabelecidas em pacientes com doenças hematológicas subjacentes (por exemplo: anemia hemolítica, anemia falciformes, talassemia).
Durante o tratamento pode ocorrer aumento dose-dependente de grau moderado da contagem plaquetária (dentro do nível normal), o qual regride durante o curso do tratamento. Além disso, o desenvolvimento de trombocitemia (aumento de plaquetas) acima do nível normal foi relatado. Recomenda-se que a contagem plaquetária seja regularmente monitorada durante as primeiras 8 semanas de tratamento.
Outras causas de anemia (deficiência de ferro, folato ou vitamina B12, intoxicação por alumínio, infecção ou inflamação, perda de sangue, hemólise e fibrose da medula óssea de qualquer origem) devem ser avaliadas e tratadas antes do início do tratamento com Eprex e quando houver aumento da dose. Na maioria dos casos, as concentrações séricas de ferritina diminuem simultaneamente ao aumento do volume globular. A fim de garantir uma resposta ótima à Eprex, os estoques de ferro devem ser adequados e suplementação de ferro deve ser administrada se necessário.
Para pacientes com cirurgia ortopédica eletiva de grande porte, a suplementação de ferro (ferro elementar 200 mg/dia por via oral) deve ser realizada durante o curso de tratamento com alfaepoetina. Se possível, a suplementação deve ser iniciada antes do início do tratamento com alfaepoetina para alcançar estoques adequados de ferro.
Eprex deve ser usado com cautela se você apresentar porfiria.
Agentes estimulantes de eritropoese (ESAs) não são necessariamente equivalentes. Portanto, somente mude de agente estimulante da eritropoese (como Eprex) para outro agente com autorização de um médico.
Pacientes com insuficiência renal
Se você apresentar insuficiência renal crônica, e é tratado com Eprex, os níveis de hemoglobina devem ser determinados regularmente até a sua estabilização e periodicamente após esta.
Pacientes com insuficiência renal crônica e resposta insuficiente da hemoglobina ao tratamento com ESAs podem estar sob maior risco de eventos cardiovasculares e mortalidade do que outros pacientes.
Tromboses do “shunt” ocorreram em pacientes em hemodiálise, especialmente naqueles com tendência à hipotensão (pressão baixa) ou cuja fístula arteriovenosa exibe complicações (por exemplo, estenoses, aneurismas, etc.). A revisão precoce do “shunt” e a profilaxia para trombose com ácido acetilsalicílico, por exemplo, é recomendada nestes pacientes.
Os eletrólitos séricos devem ser monitorados se você apresentar insuficiência renal crônica. Se for detectado nível sérico de potássio elevado ou em elevação, além do tratamento apropriado da hiperpotassemia (excesso de potássio), deve-se considerar a interrupção da administração da alfaepoetina até a correção do nível sérico de potássio.
Como resultado do aumento do volume globular, os pacientes sob hemodiálise frequentemente requerem aumento da dose de heparina durante a diálise. Se a heparinização não é adequada pode ocorrer oclusão (fechamento) do sistema de diálise.
Aplasia Pura de Células Vermelhas mediada por anticorpos
A ocorrência de aplasia pura de células vermelhas mediada por anticorpos foi raramente relatada depois de meses a anos de administração subcutânea da alfaepoetina no tratamento de pacientes com insuficiência renal crônica.
Os casos também foram raros em pacientes com hepatite C tratados com interferon e ribavirina que usaram ESAs concomitantemente. Os ESAs não são aprovados para tratamento da anemia associada à hepatite C.
Pacientes com insuficiência renal crônica tratados com alfaepoetina por via subcutânea devem ser monitorados regularmente para perda da eficácia, definida como ausência ou redução da resposta ao tratamento com alfaepoetina em pacientes que responderam previamente a este tipo de tratamento.
Isto é caracterizado por uma diminuição persistente da hemoglobina, apesar do aumento da dose de alfaepoetina.
Se houver suspeita de Aplasia Pura de Células Vermelhas, a terapia com Eprex deve ser imediatamente interrompida. Nenhuma outra terapia com ESAs deve ser iniciada devido ao risco de reação cruzada. Terapia apropriada, como transfusões de sangue, pode ser administrada, quando indicado.
Pacientes com câncer
Os níveis de hemoglobina devem ser determinados regularmente se você estiver com câncer e está recebendo Eprex, até a sua estabilização e depois periodicamente.
Há uma preocupação que os ESAs possam estimular o crescimento de tumores.
A decisão de administrar tratamento com eritropoetina recombinante deve ser baseada na avaliação do risco/benefício com participação do próprio paciente.
Em pacientes com câncer recebendo quimioterapia, deve-se levar em consideração uma demora de 2-3 semanas entre a administração de eritropoetina e o aparecimento de glóbulos vermelhos induzidos pela eritropoetina ao avaliar se o tratamento com alfaepoetina é adequado (em particular em pacientes sob risco de transfusão).
Se você apresentar uma reação grave na pele, uma erupção cutânea que pode ser grave, cobrir todo o seu corpo e também incluir bolhas ou áreas de pele descamada, pare de usar Eprex e informe ao seu médico ou procure ajuda médica imediatamente.
Pacientes infectados com HIV
Se os pacientes infectados com HIV não apresentarem resposta ou não mantiverem a resposta à Eprex, outras etiologias, incluindo anemia ferropriva, devem ser consideradas e avaliadas.
Pacientes adultos em pré-operatório em programa de pré-doação de sangue autólogo
Todas as advertências e precauções associadas aos programas de doação de sangue autólogo, especialmente reposição rotineira de volume, devem ser respeitadas caso você esteja recebendo Eprex.
Pacientes adultos em pré-operatório (sem participar do programa de doação de sangue autólogo)
Se você for submetido a cirurgia ortopédica eletiva de grande porte, você deve receber profilaxia antitrombótica adequada, uma vez que eventos trombóticos e vasculares podem ocorrer em pacientes cirúrgicos, especialmente naqueles com doença cardiovascular subjacente. Além disso, recomenda-se precaução especial caso você apresente predisposição ao desenvolvimento de trombose venosa profunda.
Em pacientes com nível de base de hemoglobina > 13 g/dL, a possibilidade do tratamento com Eprex estar associado com aumento do risco de eventos trombóticos/vasculares após a cirurgia não pode ser excluída. Portanto, a alfaepoetina não deve ser usada em pacientes com nível basal de hemoglobina > 13 g/dL.
O uso de Eprex não é recomendado em pacientes em pré-operatório com valores basais de hemoglobina superiores a 13 g/dL.
Interações Medicamentosas
Embora Eprex normalmente não reaja com outros medicamentos, informe ao seu médico se você estiver usando ou tenha recentemente usado alguma outra medicação.
Se você estiver tomando um fármaco (medicamento) conhecido por ciclosporina (para suprimir seu sistema imune após um transplante), seu médico deve solicitar testes sanguíneos especiais para verificar os níveis de ciclosporina enquanto você estiver utilizando Eprex.
A ação de Eprex poderá ser potencializada pela administração terapêutica simultânea de um agente hematínico, como o sulfato ferroso, quando houver um estado deficitário de precursores da hemoglobina.
Eprex deve ser utilizado sozinho. Não deve ser misturado a outros líquidos para a injeção.
Efeitos sobre a capacidade de dirigir veículos e utilizar máquinas
Não foram conduzidos estudos para avaliar os efeitos de Eprex sobre a capacidade de dirigir e operar máquinas.
Uso em idosos, crianças e outros grupos de risco
A segurança de Eprex não foi estabelecida em pacientes com disfunção hepática, pois, devido ao reduzido metabolismo, esses pacientes podem apresentar aumento da eritropoese.
Em pacientes com insuficiência renal crônica e doença cardíaca isquêmica clinicamente evidente ou insuficiência cardíaca congestiva a porcentagem de manutenção da hemoglobina não deve exceder o limite superior da concentração alvo, conforme recomendado.
A seleção e o ajuste da dose devem ser individualizados em pacientes idosos, a fim de atingir e manter a faixa de concentração da hemoglobina.
Gravidez, Amamentação e Fertilidade
Eprex não é recomendado em pacientes grávidas, amamentando, ou que desejam engravidar.
O efeito de Eprex na fertilidade humana não foi estudado.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Este medicamento pode causar doping.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como qualquer outro medicamento, Eprex pode causar efeitos adversos indesejáveis.
A reação adversa mais frequente durante o tratamento com alfaepoetina é o aumento da pressão arterial que pode precisar de tratamento com medicamentos, ou ajuste da dose dos medicamentos que você já esteja usando para pressão alta. Seu médico irá monitorar regularmente sua pressão arterial enquanto você estiver usando Eprex.
As reações adversas mais frequentes em estudos clínicos com a alfaepoetina são diarreia, náusea, vômito, febre e dor de cabeça. Sintomas gripais (como dor de cabeça, dor pelo corpo, dor nas articulações, fraqueza, cansaço, vertigem e calafrios) podem ocorrer principalmente no início do tratamento.
Se você apresentar estes sintomas durante injeção intravenosa de Eprex, diminuir a velocidade da injeção pode ajudar a evitá-los.
Reações de hipersensibilidade (alergia), incluindo erupção cutânea (inclusive urticária), reação anafilática e angioedema (acúmulo de fluido sob a pele das pálpebras) foram relatadas.
Incidência aumentada de eventos trombóticos vasculares (obstrução de vasos) foi observada em pacientes recebendo agentes eritropoiéticos incluindo a alfaepoetina.
Se você apresentar dor de cabeça, particularmente súbita e lancinante enxaqueca, começar a se sentir confuso ou tiver convulsão, informe ao seu médico imediatamente. Estes podem ser sinais de um aumento repentino na pressão arterial necessitando de um tratamento médico imediato.
Experiência de estudos clínicos
Dados de pós-comercialização
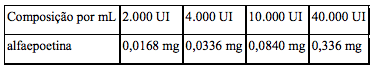
Composição
Veja a composição por mL na tabela a seguir:
 Excipientes: cloreto de sódio, fosfato de sódio monobásico di-hidratado, fosfato de sódio dibásico di-hidratado, glicina, polissorbato 80, água para injetáveis.
Excipientes: cloreto de sódio, fosfato de sódio monobásico di-hidratado, fosfato de sódio dibásico di-hidratado, glicina, polissorbato 80, água para injetáveis.
Superdosagem
A margem de segurança terapêutica de Eprex é muito ampla.
A superdose por alfaepoetina pode produzir efeitos que são derivados dos efeitos farmacológicos do hormônio. Flebotomia pode ser realizada na ocorrência de níveis excessivamente altos de hemoglobina. Deve-se tomar cuidados adicionais de suporte de acordo com o necessário.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível.
Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Embora Alfaepoetina (substância ativa) normalmente não reaja com outros medicamentos, informe seu médico se você estiver usando ou tenha recentemente usado alguma outra medicação.
Se você estiver tomando um fármaco (medicamento) conhecido por ciclosporina (para suprimir seu sistema imune após um transplante), seu médico deve solicitar testes sanguíneos especiais para verificar os níveis de ciclosporina enquanto você estiver utilizando Alfaepoetina (substância ativa).
Ação da Substância
Resultados de eficácia
2.000 UI e 4.000 UI
Foram realizados dois estudos clínicos para a avaliação do efeito terapêutico e da farmacocinética do uso intravenoso e subcutâneo da Alfaepoetina (substância ativa) no tratamento da anemia associada à insuficiência renal crônica. O primeiro estudo teve como objetivo caracterizar a farmacocinética do medicamento, avaliar seu efeito terapêutico no tratamento da insuficiência renal crônica terminal (IRCT) e determinar a tolerância e surgimento de eventos adversos pela sua administração. Para o cumprimento desses objetivos, realizou-se um ensaio clínico fase I-IIa, prospectivo, aberto, com um único grupo submetido ao tratamento durante 16 semanas. A dose inicial utilizada foi de 50 UI/kg, três vezes por semana, por via intravenosa.
Foram selecionados 25 pacientes com IRCT em hemodiálise ou diálise peritoneal, com diagnóstico de anemia com valores de hematócrito ? 28% e de hemoglobina ? 9,5 g/dl e idade entre 18 e 70 anos. Para avaliação da farmacocinética, as variáveis primárias analisadas foram t½, AUC, MRT, Vc, CL, Cmáx e, para avaliação de seu efeito terapêutico, os níveis de hematócrito, hemoglobina (semanal) e número de transfusões de sangue durante o tratamento. A avaliação da tolerância dos efeitos adversos foi verificada através de exames laboratoriais, parâmetros bioquímicos e clínicos.
Os resultados encontrados mostraram que 92% dos pacientes tratados com a Alfaepoetina (substância ativa) alcançaram o hematócrito alvo em 12 semanas de tratamento. Obteve-se um aumento global médio de hematócrito de 9,5% e de hemoglobina de 2,7 g/dl nas 16 semanas de tratamento. Não houve necessidade de transfusões por anemia associada a IRCT, a não ser para reposição sangüínea por perda aguda em virtude de cirurgia ou acidente dialítico. Houve reações adversas moderadas, sendo que o aumento da hipertensão arterial preexistente foi controlado em todos os casos com aumento das doses dos fármacos anti-hipertensivos. As características farmacocinéticas deste produto foram similares às reportadas para moléculas comerciais de Alfaepoetina (substância ativa).
O segundo estudo teve como objetivo avaliar a segurança e eficácia clínica da administração por via subcutânea do medicamento no tratamento da anemia associada à insuficiência renal crônica. Foi realizado um estudo clínico em 24 pacientes com insuficiência renal crônica terminal em hemodiálise ou diálise peritoneal, com diagnóstico de anemia com valores de hematócrito ? 28% e idade superior a 18 anos. A dose inicial utilizada foi de 20 UI/kg, três vezes por semana, por via subcutânea após sessão de hemodiálise. Para a avaliação do efeito terapêutico, as variáveis analisadas foram hematócrito, hemoglobina e número de transfusões de sangue durante o tratamento. A avaliação da tolerância dos efeitos adversos foi verificada através de registro em banco de dados.
Os resultados encontrados mostraram que 88,88% dos pacientes tratados com Alfaepoetina (substância ativa) alcançaram o hematócrito alvo (30 - 36%) durante as 12 semanas de tratamento. As necessidades transfusionais se reduziram de 94% antes do uso do medicamento a 14% depois de 12 semanas. Houve reações adversas moderadas, sendo que o aumento da hipertensão arterial preexistente foi controlado em todos os casos com aumento das doses dos fármacos anti-hipertensivos.
Foi realizado um estudo fase IV, multicêntrico, aberto, com o objetivo de avaliar efetividade e segurança do uso da Alfaepoetina (substância ativa), na correção de anemia observada no curso de quimio ou radioterapia, em pacientes oncológicos pediátricos (faixa etária entre 1 e 17 anos). Foram incluídos 157 pacientes, tratados por um período de 8 semanas, com as apresentações de 2.000 UI e 4.000 UI, na posologia semanal de 600 UI/kg, via endovenosa, ou 150 UI/kg, 3 vezes por semana, por via subcutânea. Admitiu-se um incremento até 900 UI/kg semanal, via endovenosa ou 300 UI/kg, 3 vezes por semana, via subcutânea, na quarta semana de tratamento, caso não houvesse aumento nos níveis de hemoglobina (Hb) em pelo menos 1,0 g/dl, com relação ao valor inicial. Valores de Hb e hematócrito (Ht) foram analisados, no momento basal e nas semanas 4 e 8 de tratamento, e a ocorrência de eventos adversos foi mensurada. Dos 157 pacientes incluídos, 12 interromperam o tratamento antes do final do estudo por apresentarem níveis de Hb ? 14,0 g/dl. Estes foram considerados como êxito terapêutico e incluídos na avaliação da resposta terapêutica (desfecho primário), onde o denominador considerado constituiu-se por um total de 125 pacientes.
Observou-se um aumento ? 1,5 g/dl nos valores de Hb (com relação aos valores basais) em 68,8% dos pacientes (IC 95%: 60,7 – 76,9%). O desfecho primário de efetividade também foi avaliado de acordo com a via de administração do medicamento. O incremento de 1,5 g/dl no valor de hemoglobina, na 8ª semana de tratamento, comparativamente aos valores iniciais, ocorreu em 61,4% (IC 95%: 50,0 – 72,8 %) dos pacientes que receberam o medicamento por via endovenosa e em 78,2% (IC 95%: 67,3 – 89,1%) dos pacientes que receberam o medicamento por via subcutânea. Com relação à necessidade de transfusão (desfecho secundário), a proporção de pacientes que receberam transfusão, no início do estudo, era igual a 58%. Após 8 semanas de tratamento, este percentual caiu para 42,7%.
10.000UI
Foi realizado um estudo fase IV, multicêntrico, aberto, com o objetivo de avaliar a efetividade e a segurança do uso da Alfaepoetina (substância ativa), na correção da anemia no decorrer do tratamento por quimio e/ou radioterapia. Foram incluídos 338 pacientes adultos (maiores de 18 anos), os quais receberam como esquema de tratamento a administração de 10.000 UI de Alfaepoetina (substância ativa), 3 vezes por semana durante 8 semanas, formulação de 10.000UI, por via subcutânea, avaliando-se a efetividade através da determinação dos valores de hemoglobina e hematócrito. Foram incluídos pacientes com valor de hemoglobina inferior a 10,0g/dl.
Dos 338 pacientes tratados, 81 interromperam o tratamento antes do final do estudo por apresentarem níveis de hemoglobina iguais ou superiores a 12,0g/dl. Esses pacientes foram considerados com êxito terapêutico e, incluídos juntamente aos 118 na avaliação da resposta terapêutica. Logo, o denominador da avaliação da resposta (desfecho primário) constituiu-se por um total de 199 pacientes. O numerador foi então calculado somando-se os 81 pacientes que alcançaram os níveis de hemoglobina iguais ou superiores a 12,0g/dl aos 74 pacientes que atingiram o critério de aumento em 1,5g/dl, na 8ª semana de tratamento. Esse cálculo chegou ao percentual de resposta igual a 77,9% (IC95%: 71,9-83,9%). Em relação ao desfecho secundário, foi observada uma diminuição da necessidade de transfusão nos pacientes tratados, passando de 37,3% (avaliação inicial) a 5,9% (8ª semana de tratamento).
Características farmacológicas
A Alfaepoetina (substância ativa) é uma glicoproteína produzida pelo rim, mais precisamente pelas células adjacentes aos túbulos proximais renais; sua produção é estimulada por hipóxia. Ela atua como fator hormonal de estimulação mitótica e de diferenciação, aumentando a formação de eritrócitos maduros a partir das células progenitoras eritróides. A Alfaepoetina (substância ativa) contém 165 aminoácidos e é obtida por tecnologia de DNA recombinante. Possui um peso molecular de 34.000 Dalton e é produzida em células CHO (células de ovário de hamster chinês) nas quais o gene da Alfaepoetina (substância ativa) humana foi inserido. O produto contém uma sequência de aminoácidos idêntica à da Alfaepoetina (substância ativa) natural.
2.000 UI e 4.000 UI
T½ = 5,21 h.
CL = 0.47 l/h.
AUC = 8967,42 mUI*h/ml.
Vc = 3,24 l.
MRT = 7,58 h.
Co = 1766,12 mUI/ml.
10.000 UI
A anemia é freqüentemente um sintoma concomitante em pacientes com câncer. A origem depende de uma combinação de fatores, onde os efeitos tóxicos diretos dos agentes quimioterápicos utilizados desempenham um papel importante. Nestes pacientes, a resposta ao tratamento com Alfaepoetina (substância ativa) também depende do nível endógeno da Alfaepoetina (substância ativa). Pacientes com nível endógeno superior a 200mUI/mL não respondem ao tratamento.
Cuidados de Armazenamento
Conservar sob refrigeração (entre 2ºC e 8ºC). Proteger da luz. Não congelar. Não agitar.
Se você utiliza Eprex em casa, é importante considerar os seguintes pontos:
- Eprex seringa preenchida deve ser conservado na geladeira. Contudo, não guardar no congelador e nem no “freezer”.
- Conservar Eprex na embalagem original até o momento de utilizá-lo.
- Antes de usar Eprex seringa preenchida, deixá-lo a temperatura ambiente por 15 a 30 minutos. Nunca deixe Eprex em temperatura ambiente por mais de 60 minutos antes da aplicação da injeção, nem deixe o medicamento exposto ao sol.
- Nunca aqueça Eprex.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico
Eprex é uma solução injetável transparente e translúcida.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS- 1.1236.3337
Farm. Resp.:
Marcos R. Pereira - CRF/SP nº 12.304
Registrado por:
Janssen-Cilag Farmacêutica Ltda.
Avenida Presidente Juscelino Kubitschek, 2041, São Paulo – SP
CNPJ 51.780.468/0001-87
Fabricado por:
Cilag AG
Schaffhausen - Suíça
Importado por:
Janssen-Cilag Farmacêutica Ltda.
Rodovia Presidente Dutra, km 154
São José dos Campos – SP
CNPJ 51.780.468/0002-68
Venda sob prescrição médica.
informações complementares
| Fabricante |
| JANSSEN-CILAG |
| Princípio ativo |
| Alfaepoetina |
| Categoria do medicamento |
| Medicamentos Especiais |