para o que é indicado e para que serve?
Para que serve o Ibrance Ibrance é indicado para o tratamento do câncer de mama avançado ou metastático HR (receptor hormonal) positivo e HER2 (receptor 2 do fator de crescimento epidérmico humano) negativo, em combinação com terapia endócrina: Com letrozol como terapia endócrina inicial em mulheres na pós-menopausa; Com fulvestranto em mulheres que receberam terapia prévia.Continue lendo...
ofertas de
Ibrance 75Mg 21 Cápsulas
ofertas de Ibrance 75Mg 21 Cápsulas
R$ 6.782,13
R$ 6.991,89
R$ 8.100,00
R$ 8.150,00
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve o Ibrance
Ibrance é indicado para o tratamento do câncer de mama avançado ou metastático HR (receptor hormonal) positivo e HER2 (receptor 2 do fator de crescimento epidérmico humano) negativo, em combinação com terapia endócrina:
- Com letrozol como terapia endócrina inicial em mulheres na pós-menopausa;
- Com fulvestranto em mulheres que receberam terapia prévia.
As informações sobre letrozol e fulvestranto podem ser encontradas na bula dos produtos. Seu médico também poderá lhe fornecer essas informações.
Como o Ibrance funciona?
Contraindicação do Ibrance
O uso de Ibrance é contraindicado em pacientes com alergia conhecida ao palbociclibe ou a qualquer um dos componentes da fórmula.
Como usar o Ibrance
Ibrance deve ser recomendado a você por profissionais de saúde com experiência no tratamento de câncer de mama. Sempre use Ibrance de acordo com as recomendações de seu médico. Não tome mais Ibrance além do que foi prescrito pelo seu médico.
A dose recomendada de Ibrance é uma cápsula de 125 mg, por boca, uma vez ao dia, durante 21 dias consecutivos, seguido por sete dias sem tratamento (esquema 3/1) para compor um ciclo completo de 28 dias.
Quando palbociclibe for administrado em associação com letrozol, a dose recomendada de letrozol é de 2,5 mg, por boca, uma vez ao dia, todos os dias, durante o ciclo de 28 dias.
Quando palbociclibe for administrado em associação com fulvestranto, a dose recomendada de fulvestranto é de 500 mg, administrada por via intramuscular, nos dias 1, 15, 29 e, depois, uma vez ao mês.
Ibrance deve ser tomado com alimentos, e preferencialmente no mesmo horário, todos os dias. Ibrance não deve ser administrado com toranja (grapefruit) ou suco de grapefruit.
É importante que você informe o seu médico como você está sentindo durante o seu tratamento com Ibrance.
Dependendo de sua resposta ao tratamento, seu médico poderá solicitar a alteração das doses de Ibrance ou até a interrupção do tratamento.
Insuficiência hepática
Nenhum ajuste de dose é necessário para pacientes com distúrbios do fígado (insuficiência hepática) leves ou moderados. Caso você possua algum distúrbio severo do fígado, seu médico poderá prescrever uma dose reduzida de Ibrance.
Insuficiência renal
Nenhum ajuste de dose é necessário para pacientes com distúrbios do rim (insuficiência renal) leves, moderados ou severos. Ibrance não foi avaliado em pacientes que fazem tratamento com hemodiálise.
Pacientes pediátricos
A segurança e a eficácia do palbociclibe em crianças não foram estabelecidas.
Pacientes idosos
Não é necessário ajuste na dose inicial.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que devo fazer quando eu me esquecer de usar o Ibrance?
Precauções do Ibrance
Este medicamento foi prescrito somente para você. Não compartilhe Ibrance com ninguém, mesmo se eles apresentarem sintomas semelhantes aos seus.
Neutropenia (diminuição de um tipo de células de defesa no sangue: neutrófilos)
A diminuição das células de defesa no sangue, conhecidas como neutrófilos foi o evento adverso mais frequentemente apresentado pelos pacientes que participaram dos estudos clínicos de Ibrance. Esta diminuição do número de neutrófilos (células de defesa) no sangue ocorre em média a partir do 15º dia de tratamento. Foi infrequente (?1%) o relato de diminuição de neutrófilos no sangue associado a um quadro de febre (conhecido como neutropenia febril) pelos pacientes que participaram dos estudos clínicos de Ibrance.
Deve-se monitorar o hemograma (exame de sangue) antes do início da terapia com Ibrance e no início de cada ciclo, bem como no dia 15 dos dois primeiros ciclos e conforme indicação de seu médico.
De acordo com o grau de neutropenia apresentado, seu médico poderá optar por interrupção ou a redução da dose ou o adiamento do início dos ciclos de tratamento até que ocorra a recuperação dos níveis de neutrófilos no sangue.
Infecções
Como Ibrance pode causar diminuição das células de defesa do sangue (leucócitos, neutrófilos), os pacientes que fizerem uso da medicação podem ter predisposição a infecções.
Portanto, pacientes que fizerem uso de Ibrance devem relatar, imediatamente ao seu médico, quaisquer episódios de febre.
Ablação (extirpação) / supressão dos ovários em mulheres em pré/perimenopausa
Ablação (extirpação) ovariana ou sua supressão com uso de medicamentos (conhecidos como agonistas do receptor LHRH) é mandatória quando mulheres na pre/perimenopausa tomam Ibrance em combinação com um inibidor de aromatase, devido ao mecanismo de ação dos inibidores de aromatase. Palbociclibe em combinação com fulvestranto em mulheres na pre/perimenopausa só foi estudado em combinação com medicamento supressor do ovário como os agonistas do receptor LHRH.
Fertilidade, gravidez e lactação
Nos estudos clínicos, não foram obtidos dados sobre o impacto de Ibrance na fertilidade de mulheres. De acordo com os achados dos estudos conduzidos em animais (pré-clinicos), a fertilidade masculina pode ser comprometida pelo tratamento com Ibrance. Os homens devem considerar a preservação (congelamento) de esperma antes do início do tratamento com Ibrance.
Se você for homem e estiver usando Ibrance, você deverá utilizar preservativo durante as relações sexuais (mesmo que tenha sido submetido a uma vasectomia bem sucedida) durante o tratamento e interrupções de dose, e por pelo menos 97 dias após a descontinuação da terapia com Ibrance.
Não há estudos adequados e bem-controlados sobre o uso de Ibrance em mulheres grávidas. Palbociclibe pode causar danos ao feto quando administrado a mulheres grávidas, baseado nos achados dos estudos conduzidos em animais e no mecanismo de ação do medicamento.
Mulheres em idade fértil que estejam recebendo este medicamento, ou os parceiros de mulheres em idade fértil que estejam recebendo este medicamento, devem usar métodos contraceptivos adequados (por exemplo, contracepção de barreira dupla, como preservativo e diafragma) durante o tratamento e, por no mínimo 21 dias (mulheres) ou 97 dias (homens) após o término do tratamento. Discuta com o seu médico sobre métodos eficazes para evitar a gravidez que melhor se adaptam a você.
Mulheres com potencial para engravidar devem fazer testes de gravidez antes do início do tratamento com Ibrance.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Não há informações sobre a presença de Ibrance no leite humano e sobre seus efeitos no bebê. Em decorrência do potencial de Ibrance provocar reações adversas graves em bebês, a amamentação deve ser descontinuada durante o tratamento com Ibrance e por 3 semanas após a última dose. Você deverá discutir com seu médico sobre interromper a amamentação ou interromper o uso de Ibrance, levando em consideração a importância do mesmo para o seu tratamento.
Efeitos na Habilidade de Dirigir e Operar Máquinas
Não foram realizados estudos sobre o efeito do Ibrance na habilidade de dirigir veículos ou operar máquinas.
Entretanto, os pacientes que apresentarem fadiga (cansaço) com o uso de Ibrance devem ter cautela ao dirigir ou operar máquinas.
Atenção: este medicamento contém lactose. Pacientes com problemas hereditários de intolerância à galactose ou outros açúcares, deficiência de lactase de Lapp ou má absorção de glucose-galactose devem consultar o médico antes de tomar este medicamento.
Reações Adversas do Ibrance
Informe ao seu médico imediatamente se você tiver algum destes sintomas:
Febre, calafrios, fraqueza, falta de ar, sangramentos ou hematomas. Estes podem ser sinais de um distúrbio sanguíneo grave.
As reações adversas ao Ibrance estão listadas a seguir:
Composição do Ibrance
Apresentações
Ibrance 75 mg, 100 mg ou 125 mg em embalagens contendo 21 cápsulas duras.
Via de administração: Uso oral.
Uso adulto.
Composição
Superdosagem do Ibrance
Não há antídoto conhecido para o palbociclibe. O tratamento para superdosagem de Ibrance deve consistir de medidas gerais de suporte.
Muitos pacientes que tomaram doses acima da dose terapêutica recomendada, fizeram isso de forma acidental.
Os eventos adversos mais comumente relatados em casos de superdosagem são aqueles relacionados à diminuição das células presentes na medula óssea, por exemplo, diminuição dos neutrófilos e anemia, que podem ser agravados ou prolongados. Em alguns casos, também foram descritos sintomas gastrointestinais (ex: náuseas e vômitos).
Procure imediatamente seu médico ou hospital se você tomar acidentalmente mais Ibrance do que o médico prescreveu. Você deve mostrar a caixa de Ibrance. Um tratamento médico pode ser necessário.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001 se você precisar de mais orientações.
Interação Medicamentosa do Ibrance
É importante que você mantenha uma lista escrita de todos os medicamentos sob prescrição médica e sem prescrição que você está tomando; bem como quaisquer produtos, tais como vitaminas, minerais ou outros suplementos dietéticos. Você deve trazer esta lista com você cada vez que você visitar o médico ou se você está internado em um hospital. Esta lista também é uma informação importante para levar com você em caso de emergências.
Como Ibrance pode ser utilizado em associação com letrozol ou fulvestranto, você deve conversar com seu médico também sobre outros medicamentos que devem ser evitados quando se toma qualquer uma dessas medicações.
Ibrance pode afetar o modo de ação de outros medicamentos.
Em particular, os seguintes produtos podem aumentar o risco de reações adversas com Ibrance:
- Amprenavir, atazanavir, delavirdina, fosamprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir e telaprevir< utilizados para tratar infecção pelo virus HIV/AIDS.
- Boceprevir e telaprevir utilizados para tratar infecção pelo vírus da Hepatite C.
- Antibacterianos claritromicina, eritromicina e telitromicina utilizados para tratar infecções causadas por bacterias.
- Itraconazol, cetoconazol, miconazol, posaconazol e voriconazol utilizados para tratar infecções causadas por fungos (ex. micoses).
- Diltiazem e mibefradil utilizados para tratar hipertensão e angina pectoris crônica.
- Nefazodona utilizada para tratar depressão.
- Conivaptan utilizado para tratar certas desordens dos eletrólitos no sangue (ex. hiponatremia - diminuição dos níveis de sódio no sangue).
- Grapefruit (toranja) e suco de grapefruit.
Ibrance pode aumentar o risco de reações adversas relacionados aos seguintes produtos:
- Quinidina geralmente utilizada para tratar problemas de ritmo cardíaco.
- Colchicina utilizada para tratar gota.
- Digoxina utilizada para tratar insuficiência cardíaca e problemas de ritmos cardíacos.
- Pravastatina e rosuvastatina utilizadas para tratar altos níveis de colesterol.
- Sulfasalazina utilizada para tratar artrite reumatoide.
- Alfentanila utilizada para anestesia em cirurgia; fentanila utilizada em pré-procedimentos como alivio de dor assim como anestésico.
- Ciclosporina, everolimo, tacrolimo e sirolimo utilizados na prevenção de rejeição em transplante de órgãos.
- Diidroergotamina e ergotamina utilizadas para tratar enxaquecas.
- Pimozida utilizada para tratar esquizofrenia e psicose crônica.
- Metformina para tratar diabetes.
Os seguintes produtos podem reduzir a eficácia de Ibrance:
- Carbamazepina, felbamato, fenobarbital, fenitoína e primidona utilizadas para parar convulsões e ataques.
- Rifabutina, rifampina e rifapentina utilizadas para tratar tuberculose.
- Nevirapina utilizada para tratar infecção por vírus HIV/AIDS.
- Enzalutamida utilizada para tratar certos tipos de câncer.
- Erva de São João, utilizada para tratar depressão e ansiedade leves.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Ação da Substância Ibrance
Resultados de Eficácia
Estudo 1 - Estudo randomizado de Fase 1/2 de Palbociclibe (substância ativa) em combinação com letrozol (PALOMA-1)
A eficácia de Palbociclibe (substância ativa) foi avaliada em um estudo randomizado, aberto, multicêntrico de Palbociclibe (substância ativa) mais letrozol versus letrozol isolado, com mulheres pós-menopausa com câncer de mama avançado ER-positivo e HER2-negativo que não receberam tratamento sistêmico prévio para a doença avançada (PALOMA-1).
O estudo foi composto por uma parte de Fase 1 limitada (N = 12), projetada para confirmar a segurança e a tolerabilidade da combinação de Palbociclibe (substância ativa) mais letrozol, seguida por uma parte randomizada de Fase 2 (N = 165), destinada a avaliar a eficácia e a segurança de Palbociclibe (substância ativa) em combinação com letrozol, em comparação com letrozol isolado, no tratamento de primeira linha de mulheres pós-menopausa com câncer de mama avançado ER-positivo, HER2-negativo.
A randomização foi estratificada de acordo com o local da doença (visceral versus somente óssea versus outros) e intervalo livre de doença (>12 meses desde o final do tratamento adjuvante até a recorrência da doença versus ?12 meses do final do tratamento adjuvante até a recorrência da doença ou doença avançada de novo).
As características clínicas e demográficas das pacientes na avaliação inicial eram, em geral, equilibradas entre os braços do estudo no que se refere à idade, raça, locais da doença, estágio e terapias prévias. A Tabela 1, abaixo, apresenta um resumo detalhado das características clínicas e demográficas na avaliação inicial.
Tabela 1. Resumo das características clínicas e demográficas na avaliação inicial por tipo de tratamento – Estudo 1 (população com intenção de tratar)
| Parâmetro | Palbociclibe (substância ativa) mais letrozol (N = 84) | Letrozol (N = 81) |
| Idade (anos) | ||
| Mediana (mín., máx.) | 62,5 (41, 89) | 64,0 (38, 84) |
| <65 [n (%)] | 47 (56,0) | 42 (51,9) |
| ?65 [n (%)] | 37 (44,0) | 39 (48,1) |
| Status de desempenho ECOG [n (%)] | ||
| 0 | 46 (54,8) | 45 (55,6) |
| 1 | 38 (45,2) | 36 (44,4) |
| Local da doença* [n (%)] | ||
| Visceral | 39 (46,4) | 40 (49,4) |
| Óssea apenas | 17 (20,2) | 14 (17,3) |
| Outros | 28 (33,3) | 27 (33,3) |
| Intervalo livre de doença* [n (%)] | ||
| >12 meses desde o final do tratamento adjuvante até a recorrência da doença | 37 (44,1) | 36 (44,4) |
| ?12 meses desde o final do tratamento adjuvante até a recorrência da doença ou doença avançada de novo | 47 (56,0) | 45 (55,6) |
| Terapia sistêmica prévia [n (%)] | ||
| Não | 44 (52,4) | 37 (45,7) |
| Sim | 40 (47,6) | 44 (54,3) |
| Quimioterapia [n (%)] | 34 (40,5) | 37 (45,7) |
| Antraciclina | 26 (31,0) | 25 (30,9) |
| Taxano | 12 (14,3) | 14 (17,3) |
| Outros | 34 (40,5) | 37 (45,7) |
| Terapia anti-hormonal [n (%)] | 27 (32,1) | 28 (34,6) |
| Tamoxifeno | 24 (28,6) | 24 (29,6) |
| Inibidor de aromatase | 14 (16,7) | 12 (14,8) |
| Anastrozol | 8 (9,5) | 11 (13,6) |
| Exemestano | 4 (4,8) | 2 (2,5) |
| Letrozol | 2 (2,4) | 1 (1,2) |
| Outros | 2 (2,4) | 0 |
*Com base na randomização.
Abreviações: ECOG (Eastern Cooperative Oncology Group) = grupo de estudo de oncologia do leste; Máx. = máximo; Mín. = mínimo; N/n = número de pacientes.
O endpoint primário do estudo, a sobrevida livre de progressão (PFS - progression-free survival), foi avaliado pelo investigador de acordo com o critério de avaliação de resposta em tumores sólidos (RECIST - Response Evaluation Criteria in Solid Tumors) versão 1.0.
A PFS mediana (mPFS) das pacientes no braço de Palbociclibe (substância ativa) mais letrozol foi de 20,2 meses (intervalo de confiança de 95% [IC]: 13,8; -27,5) e 10,2 meses (IC de 95%: 5,7; -12,6) para pacientes do braço de letrozol isolado. A razão de riscos observada (HR – hazard ratio) foi de 0,488 (IC de 95%: 0,319; -0,748) em favor de Palbociclibe (substância ativa) mais letrozol, com um teste log-rank unilateral estratificado de valor p = 0,0004.
Os resultados da eficácia primária foram obtidos na análise de PFS final de todas as pacientes randomizadas para Estudo 1 (vide Figura 1 abaixo).
Figura 1. Curvas de Kaplan-Meier para Sobrevida Livre de progressão (Fase 2, Avaliação do Investigador, População com Intenção de Tratar)
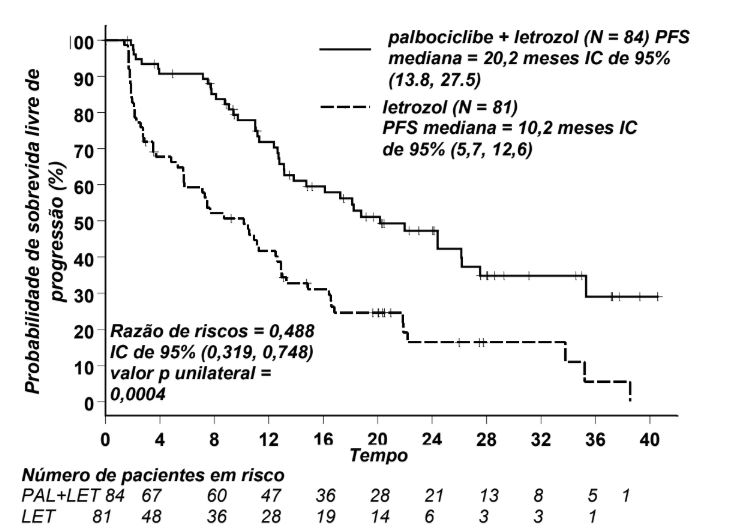
IC = intervalo de confiança; LET = letrozol; N = número de pacientes; PAL = Palbociclibe (substância ativa); PFS = sobrevida livre de progressão.
Estudo 2 - Estudo randomizado de Fase 3 de Palbociclibe (substância ativa) em combinação com letrozol (PALOMA-2)5,6
A eficácia de Palbociclibe (substância ativa) em combinação com letrozol, versus placebo mais letrozol, foi avaliada em um estudo internacional, randomizado, duplo-cego, controlado por placebo, multicêntrico, de grupo paralelo, conduzido com mulheres com câncer de mama avançado ou metastático ER-positivo, HER2-negativo (PALOMA-2), que não receberam tratamento sistêmico prévio para sua doença avançada.
Um total de 666 mulheres na pós-menopausa foram randomizadas a 2:1 para o braço de Palbociclibe (substância ativa) mais letrozol ou para o braço de placebo mais letrozol e estratificadas de acordo com o local da doença (visceral, não visceral), com o intervalo livre de doença desde o final do tratamento (neo)adjuvante até recorrência da doença (metastática de novo, ?12 meses desde o final do tratamento adjuvante até recorrência da doença, >12 meses desde o final do tratamento adjuvante até recorrência da doença) e com o tipo de tratamentos antineoplásicos (neo) adjuvantes (terapia hormonal prévia, sem terapia hormonal prévia). As pacientes com doença visceral disseminada sintomática e avançada, que correm risco de complicações potencialmente fatais em curto prazo (incluindo pacientes com efusões não controladas [pleural, pericárdica, peritoneal], linfangite pulmonar e mais de 50% de envolvimento hepático) não eram qualificáveis para inclusão no estudo.
As pacientes continuaram a receber o tratamento designado até a progressão objetiva da doença, deterioração sintomática, toxicidade intolerável, morte ou retirada de consentimento, o que ocorresse primeiro. O cruzamento entre os braços de tratamento não foi permitido.
As pacientes foram combinadas quanto a características demográficas e clínicas da doença na avaliação inicial,entre o braço de Palbociclibe (substância ativa) mais letrozol e o braço de placebo mais letrozol. A idade mediana das pacientes inscritas neste estudo era de 62 anos (faixa de variação de 28-89). 48,3% das pacientes tinham recebido quimioterapia e 56,3% receberam terapia anti-hormonal prévia no cenário neo (adjuvante) anterior a seu diagnóstico de câncer de mama avançado, ao passo que 37,2% das pacientes não receberam terapia sistêmica prévia no cenário (neo) adjuvante. A maioria das pacientes (97,4%) tinha doença metastática na avaliação inicial; 22,7% das pacientes tinham apenas doença óssea e 49,2% das pacientes tinham doença visceral.
O endpoint primário do estudo (PFS) foi avaliado pelo investigador de acordo com o RECIST, versão 1.1. Os endpoints secundários de eficácia incluíram taxa de resposta objetiva (ORR), duração da resposta (DR), taxa de benefício clínico (CBR), sobrevida global (OS), segurança, pontuações EQ-5D e mudança na qualidade de vida (QoL) relacionada à saúde avaliada usando o questionário FACT-B.
O estudo cumpriu o objetivo primário de melhora do PFS. A HR estimada foi de 0,576 (IC de 95%: 0,463; 0,718) em favor de Palbociclibe (substância ativa) mais letrozol, com um teste log-rank unilateral estratificado de valor p <0,000001. A mPFS foi de 24,8 meses (IC de 95%: 22,1; não estimável [NE]), para pacientes no braço de Palbociclibe (substância ativa) mais letrozol, e de 14,5 meses (IC de 95%: 12,9; 17,1), no braço de placebo mais letrozol. O efeito do tratamento da combinação no PFS também foi respaldado por uma análise independente de radiografias com uma HR estimada de 0,653 (IC de 95%: 0,505; 0,844).
Na Tabela 2, estão resumidos os dados de eficácia do estudo PALOMA-2 e a curva Kaplan-Meier para o PFS é exibida na Figura 2.
Tabela 2. Resultados de eficácia do estudo PALOMA-2 (população com intenção de tratar)
| Data de corte: 26-01-2016 | ||
| Palbociclibe (substância ativa) mais letrozol (N=444) | Placebo mais letrozol (N=222) | |
| Sobrevida livre de progressão | ||
| Avaliação do investigador, número de eventos (%) | 194 (43,7%) | 137 (61,7%) |
| Mediana [meses (IC de 95%) | 24,8 (22,1; NE) | 14,5 (12,9; 17,1) |
| Razão de riscos (IC de 95%) e valor p unilateral | 0,576 (0,46; 0,72), p<0,000001 | |
| Análise radiográfica independente, número de eventos (%) | 152 (34,2%) | 96 (43,2%) |
| Mediana [meses (IC de 95%) | 30,5 (27,4; NE) | 19,3 (16,4; 30,6) |
| Razão de riscos (IC de 95%) e valor p unilateral | 0,653 (0,505; 0,84), p=0,000532 | |
| Parâmetros secundários de eficácia (avaliação do investigador) | ||
| ORR (IC de 95%) | 46,4 (41,7; 51,2) | 38,3 (31,9; 45,0) |
| ORR (doença mensurável) [% (IC de 95%)] | 60,7 (55,2; 65,9) | 49,1 (41,4; 56,9) |
| CBR [% (IC de 95%)] | 85,8 (82,2; 88,9) | 71,2 (64,7; 77,0) |
N = número de pacientes; IC = intervalo de confiança; NE = não estimável; TORR = taxa de resposta objetiva; CBR = taxa de benefício clínico.
Os resultados dos endpoints secundários são baseados em respostas confirmadas e não confirmadas, de acordo com o RECIST 1.1.
Figura 2. Gráfico de Kaplan-Meier para sobrevida livre de progressão (avaliação do investigador, população com intenção de tratar) – Estudo Paloma-2
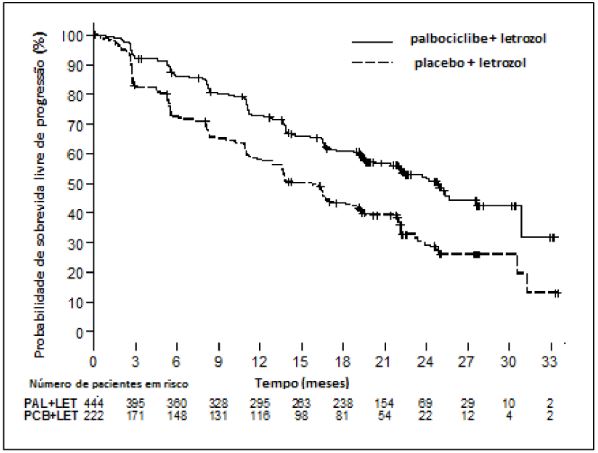
PAL=Palbociclibe (substância ativa); LET=letrozol; PCB=placebo.
Tabela 3: Resultados de eficácia em doença visceral e não visceral do estudo PALOMA-2 (população com intenção de tratar)

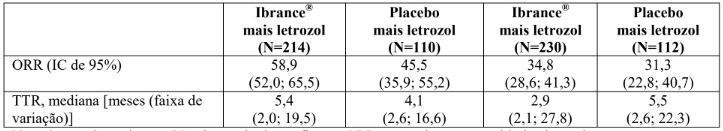
N = número de pacientes; IC = intervalo de confiança; ORR = taxa de resposta objetiva baseada em respostas confirmadas e não confirmadas de acordo com os critérios RECIST 1.1; TTR = tempo até a primeira resposta tumoral.
Uma série de análises pré-especificadas de PFS por sub-grupo foi realizada com base nas características demográficas e clínicas da doença na avaliação inicial, para investigar a consistência interna do efeito do tratamento. Observou-se uma redução no risco de progressão da doença, ou morte, no braço de Palbociclibe (substância ativa) mais letrozol em todos os subgrupos individuais de pacientes definidos por fatores de estratificação e características da avaliação inicial. Isto foi evidente em pacientes com metástase visceral (HR de 0,67 [IC de 95%: 0,50; 0,89], mPFS: 19,2 versus 12,9 meses) ou sem metástase visceral (HR de 0,48 [IC de 95%: 0,34; 0,67], mPFS: não atingida [NR] versus 16,8 meses) e pacientes apenas com doença óssea (HR de 0,36 [IC de 95%: 0,22; 0,59], mPFS: NR versus 11,2 meses) ou sem doença apenas óssea (HR de 0,65 [IC de 95%: 0,51; 0,84], mPFS: 22,2 versus 14,5 meses). De maneira similar, observou-se uma redução no risco de progressão da doença ou morte no braço de Palbociclibe (substância ativa) mais letrozol em 512 pacientes cujos tumores foram positivos para a expressão da proteína Rb por imuno-histoquímica (IHC) (HR de 0,531 [IC de 95%: 0,42, 0,68], mPFS de 24,2 meses em comparação com 13,7 meses). A redução do risco de progressão da doença ou morte a favor do braço de Palbociclibe (substância ativa) mais letrozol não foi estatisticamente significativa nos 51 pacientes cujos tumores foram negativos para a expressão da proteína Rb por IHC (HR de 0,675 [IC de 95%: 0,31, 1,48], mPFS de NR em comparação com 18,5 meses).
Uma análise do endpoint composto de tempo para deterioração (TTD), no questionário Functional Assessment of Cancer Therapy-Breast (FACT-B), definido como o tempo entre a avaliação inicial e a primeira ocorrência de diminuição de ?7 pontos nas pontuações do FACT-B, foi conduzida com base nos métodos de análise de sobrevida, usando um modelo de risco proporcional de Cox e um teste long-rank. Nenhuma diferença estatisticamente significativa no TTD foi observada nas pontuações totais do FACT-B entre o braço de Palbociclibe (substância ativa) mais letrozol e o braço de placebo mais letrozol (HR de 1,042 [IC de 95%: 0,838; 1,295]; valor unilateral p = 0,663.
Estudo 3: Estudo randomizado de Fase 3 de Palbociclibe (substância ativa) em combinação com fulvestranto
A eficácia de Palbociclibe (substância ativa) em combinação com fulvestranto versus placebo mais fulvestranto foi avaliada em um estudo internacional, randomizado, duplo-cego, multicêntrico, de grupo paralelo conduzido com mulheres com câncer de mama avançado HR-positivo, HER2-negativo, independentemente de seu status menopáusico, cuja doença progrediu após terapia endócrina prévia.
Um total de 521 mulheres em pré/pós-menopausa, cujas doenças progrediram durante, ou dentro de, 12 meses após conclusão do tratamento endócrino adjuvante ou durante, ou dentro de, 1 mês após o tratamento endócrino para doença avançada, foram randomizadas a 2:1 para o braço de Palbociclibe (substância ativa) mais fulvestranto ou para o placebo mais fulvestranto e estratificadas de acordo com sensibilidade à terapia hormonal documentada em tratamento prévio, status menopáusico ao entrar no estudo (pré/peri versus pós-menopausa) e presença de metástases viscerais. As mulheres pré/perimenopáusicas receberam gosserrelina agonista do LHRH. As pacientes com doença visceral disseminada sintomática e avançada/metastática, que correm risco de complicações potencialmente fatais em curto prazo (incluindo pacientes com efusões não controladas [pleural, pericárdica, peritoneal], linfangite pulmonar e mais de 50% de envolvimento hepático) não eram qualificáveis para inscrição no estudo.
As pacientes continuaram a receber o tratamento designado até a progressão objetiva da doença, deterioração sintomática, toxicidade intolerável, morte ou retirada de consentimento, o que ocorresse primeiro. O cruzamento entre os braços de tratamento não foi permitido.
As pacientes foram equilibradas quanto a características prognósticas e demográficas da avaliação inicial entre o braço de Palbociclibe (substância ativa) mais fulvestranto e o braço de placebo mais fulvestranto. A maioria das pacientes em cada braço de tratamento era branca, com idade média de 57 anos (variando entre 29 e 88 anos), sensibilidade documentada à terapia hormonal prévia e estava em pós-menopausa.
Aproximadamente 20% estavam em pré/perimenopausa. Todas as pacientes haviam recebido terapia sistêmica prévia e a maioria, em cada braço de tratamento, havia recebido um regime quimioterápico anterior. Mais da metade (62%) apresentou um status de desempenho do Eastern Cooperative Oncology Group (ECOG) de 0, 60% teve metástases viscerais, e 60% tinha recebido mais de 1 terapia endócrina anterior para o diagnóstico primário.
O endpoint primário do estudo (PFS) foi avaliado pelo investigador de acordo com o critério RECIST versão 1.1. Análises complementares de PFS foram baseadas em uma revisão radiológica central independente. Os endpoints secundários incluíram ORR, DOR, CBR, OS segurança, mudança na QoL e TTD. Os resultados relatados por pacientes, incluindo QoL global e dor, foram medidos usando o questionário de qualidade de vida (QLQ-C30) da Organization for Research and Treatment of Cancer (EORTC) e o questionário Breast Cancer Module (BR23).
O estudo atingiu o endpoint primário de avaliação de prolongamento da PFS avaliado pelo investigador na análise interina baseada em 82% dos eventos PFS planejados na análise final; os resultados ultrapassaram limite de eficácia Haybittle-Peto pré-especificado (? = 0,00135), demonstrando um prolongamento estatisticamente significativo na PFS e um efeito terapêutico clinicamente relevante.
A HR estimada da análise estratificada foi 0,422 (IC de 95%: 0,318; 0,560; p<0,000001 unilateral) em favor do Palbociclibe (substância ativa) mais fulvestranto.
A mPFS foi de 9,2 meses (IC de 95%:7,5), no braço de Palbociclibe (substância ativa) mais fulvestranto, e de 3,8 meses (IC de 95%: 3,5; -5,5), no braço de placebo mais fulvestranto.
Tabela 4: Resultados de eficácia – Endpoints primários (avaliação do investigador, população com intenção de tratar)
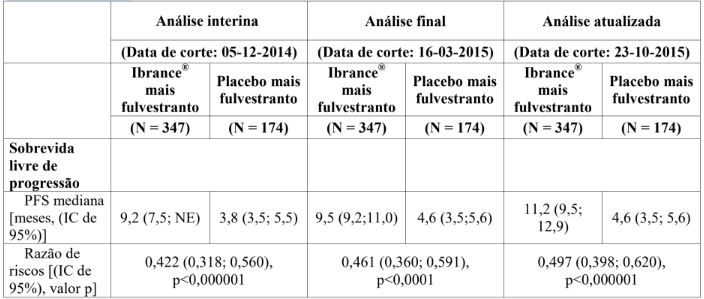
IC = intervalo de confiança; PFS = sobrevida livre de progressão.
Tabela 5: Resultados de eficácia – Endpoints secundários (avaliação do investigador, população com intenção de tratar)
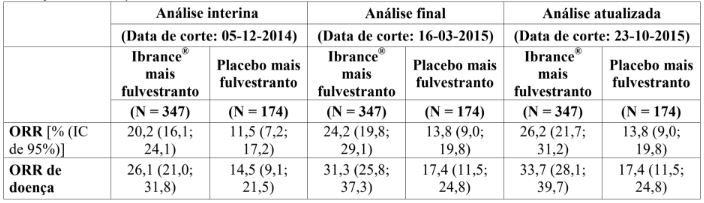
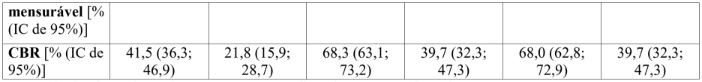
Os resultados foram baseados nas respostas confirmadas e não confirmadas.
CBR = taxa de benefício clínico; IC = intervalo de confiança; N = número de pacientes; NE = não estimável; ORR = taxa de resposta objetiva.
Figura 3. Gráfico de Kaplan-Meier para sobrevida livre de progressão (avaliação do investigador, população com intenção de tratar) - estudo PALOMA-3 (data de corte 23-10-2015)
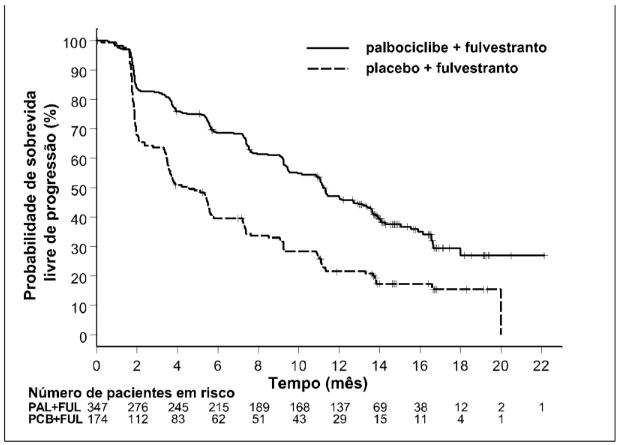
FUL = fulvestranto; PAL = Palbociclibe (substância ativa); PCB = placebo.
Tabela 6. Resultados de eficácia em doença visceral e não visceral do estudo PALOMA-3 (população com intenção de tratar)
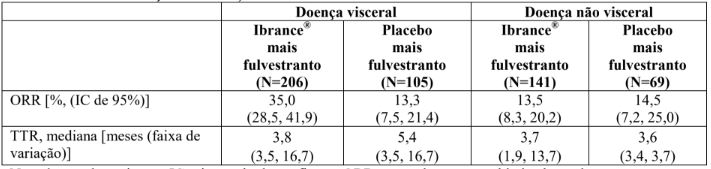
N = número de pacientes; IC = intervalo de confiança; ORR = taxa de resposta objetiva baseada em respostas confirmadas e não confirmadas, de acordo com o RECIST 1.1; TTR = tempo até a primeira resposta do tumor.
O prolongamento da PFS no braço de Palbociclibe (substância ativa) mais fulvestranto também foi demonstrado em subgrupos individuais de pacientes, o que corrobora a consistência interna de achados de benefícios de PFS do estudo, e foi corroborado por uma análise de auditoria de revisão central independente e cega (BICR) de amostra aleatória conduzida em 40,5% (N = 211) dos 521 pacientes randomizados.
As mulheres na pré/perimenopausa foram inscritas no estudo e receberam o antagonista de LHRH gosserrelina por, pelo menos, quatro semanas antes e em toda a duração do Estudo 2.
O braço de Palbociclibe (substância ativa) mais fulvestranto demonstrou benefício clínico similar na população de pacientes na pré/perimenopausa (HR = 0,435 [IC de 95%: 0,228-0,831]) e população pós-menopausa (HR = 0,409 [IC de 95%: 0,298-0,560]). De maneira similar, a mPFS para o braço de Palbociclibe (substância ativa) mais fulvestranto foi de 9,5 meses (IC de 95%: 7,2-NE) no cenário pré/perimenopausa versus 9,2 meses (IC de 95%: 7,5-NE) no cenário pós-menopausa; enquanto que a mPFS no braço de placebo mais fulvestranto foi de 5,6 meses (IC de 95%:1,8; NE) no cenário pré/perimenopausa versus 3,7 meses (IC de 95%: 3,5; -5,5) no cenário pós-menopausa. Os sintomas relatados pelas pacientes foram avaliados usando o EORTC QLQ-C30 e o EORTC QLQ-BR23. Um total de 335 pacientes no braço de Palbociclibe (substância ativa) mais fulvestranto e 166 pacientes no braço de placebo mais fulvestranto concluíram o questionário na visita de avaliação inicial e, pelo menos, em uma visita pós-avaliação inicial.
Os resultados da comparação do estado de saúde global/QoL entre o braço de Palbociclibe (substância ativa) mais fulvestranto contra o braço de fulvestranto mais placebo mostraram uma diferença estatisticamente significativa em favor do braço que contém Palbociclibe (substância ativa) mais fulvestranto comparado com o braço de placebo mais fulvestranto (-0,9 [IC de 95%: -2,5; 0,7] versus -4,0 [IC de 95%: -6,3; 1,7], respectivamente; valor p bilateral = 0,0313). Além disso, uma comparação em relação ao funcionamento emocional também mostrou uma diferença estatisticamente significativa em favor do braço de Palbociclibe (substância ativa) mais fulvestranto comparado com o braço de placebo mais fulvestranto (2,7 [IC de 95%: 1,1; 4,3] versus -1,9 [IC de 95%: -4,2; 0,5], respectivamente; valor p bilateral = 0,0016) (dados não ajustados para comparações múltiplas).
O tempo para deterioração (TTD) foi pré-definido como tempo entre a avaliação inicial e a primeira ocorrência de aumento ?10 de pontos da avaliação inicial nas pontuações de sintomas de dores. A adição de Palbociclibe (substância ativa) a fulvestranto resultou em um benefício de sintoma, retardando significativamente o TTD em pontuações de sintomas de dores comparado com o placebo mais fulvestranto (mediana de 8 meses versus 2,8 meses; HR de 0,64 [IC de 95%: 0,49; 0,85]; p<0,001).
Características Farmacológicas
Propriedades Farmacodinâmicas
O Palbociclibe (substância ativa) é administrado por via oral e é um inibidor de molécula pequena reversível e altamente seletivo das quinases dependentes de ciclina (CDK) 4 e 6. A ciclina D1 e a CDK4/6 são vias de sinalização downstream múltiplas que levam à proliferação celular. Pela inibição de CDK4/6, o Palbociclibe (substância ativa) reduziu a proliferação celular, bloqueando a progressão da célula de G1 para a fase S do ciclo celular. Testes de Palbociclibe (substância ativa) em um painel de linhagens de células de câncer de mama molecularmente perfiladas revelaram alta eficácia contra câncer de mama luminal, particularmente câncer da mama com receptor de estrogênio (ER) positivo. Análises mecanísticas revelaram que a combinação de Palbociclibe (substância ativa) com agentes antiestrogênio aumentou a reativação da proteína retinoblastoma (Rb) por meio da inibição da fosforilação do Rb, resultando em sinalização de E2F reduzida e interrupção do crescimento. A interrupção do crescimento intensificada nas linhagens de células de câncer de mama de ER-positivo tratadas com Palbociclibe (substância ativa) e agentes antiestrogênio é acompanhada pelo aumento da senescência celular, resultando em uma interrupção contínua no ciclo celular após a remoção do medicamento e aumento do tamanho da célula associado a um fenótipo senescente. Estudos in vivo com modelo de xenoenxerto do câncer da mama ER-positivo derivado de paciente (HBCx-34) demonstraram que a combinação de Palbociclibe (substância ativa) e letrozol melhorou, de maneira ainda mais pronunciada, a inibição da fosforilação do Rb, a sinalização downstream e o crescimento tumoral dose-dependente. Isso confirma a contribuição da interrupção de crescimento associada à senescência como um mecanismo associado à eficácia antitumoral da combinação Palbociclibe (substância ativa)/antagonista ER em modelos de câncer de mama ER-positivo.
Na presença ou ausência de um antiestrogênico, as células da medula óssea tratadas com Palbociclibe (substância ativa) não se tornaram senescentes e retomaram a proliferação após a retirada do Palbociclibe (substância ativa), consistente com a quiescência farmacológica. As células de câncer de mama in vitro, por outro lado, se tornaram senescentes após o tratamento com Palbociclibe (substância ativa) ou antiestrogênio com efeitos aditivos em combinação e permaneceram contidas na presença de antiestrogênio.
Propriedades Farmacocinéticas
A farmacocinética de Palbociclibe (substância ativa) foi caracterizada em pacientes com tumores sólidos que incluíam câncer de mama avançado e em indivíduos sadios.
Absorção
O tempo para Cmáx (Tmáx) de Palbociclibe (substância ativa) está geralmente entre 6 a 12 horas após administração oral. A biodisponibilidade absoluta média de Palbociclibe (substância ativa) após uma dose oral de 125 mg é de 46%. Na faixa de variação de dosagem de 25 mg a 225 mg, a AUC e Cmáx aumentam de forma proporcional à dose em geral. O estado de equilíbrio foi obtido em 8 dias após repetição da dosagem de uma vez ao dia. Com a repetição da administração de uma vez ao dia, o Palbociclibe (substância ativa) se acumula a uma razão de acúmulo mediana de 2,4 (faixa de variação de 1,5-4,2).
Distribuição
A ligação de Palbociclibe (substância ativa) a proteínas do plasma humano in vitro foi de ~85%, sem dependência de concentração numa faixa de variação de concentração de 500 ng/mL a 5.000 ng/mL. A fração média não ligada (fu) de Palbociclibe (substância ativa) no plasma humano in vivo aumentou incrementalmente com piora da função hepática. Não houve tendência obvia no fu médio de Palbociclibe (substância ativa) no plasma humano in vivo com piora da função renal. A média geométrica do volume de distribuição aparente (Vz/F) foi de 2.583 (26%) L.
Metabolismo
Estudos in vitro e in vivo indicam que Palbociclibe (substância ativa) sofre extenso metabolismo hepático em humanos. Após a administração oral de uma dose única de 125 mg de [14C]Palbociclibe (substância ativa) em humanos, as principais vias metabólicas primárias para Palbociclibe (substância ativa) envolveram oxidação e sulfonação, com acilação e glucuronidação contribuindo como vias secundárias. O Palbociclibe (substância ativa) foi a principal entidade circulante derivada de medicamento no plasma. O principal metabólito circulante foi um conjugado glucuronida de Palbociclibe (substância ativa), embora ele tenha representado somente 1,5% da dose administrada nas excreções. A maior parte do material foi excretado como metabólitos. Nas fezes, o conjugado de ácido sulfâmico do Palbociclibe (substância ativa) foi o principal componente relacionado ao medicamento, responsável por 25,8% da dose administrada. Estudos in vitro com hepatócitos humanos, frações cistosólicas e frações S9 hepáticas, e enzimas sulfotransferase recombinantes (SULT) indicaram que CYP3A e SULT2A1 são os principais envolvidos no metabolismo do Palbociclibe (substância ativa).
Eliminação
A média geométrica do clearance oral aparente (CL/F) do Palbociclibe (substância ativa) foi 63,08 L/h e a meia-vida de eliminação plasmática média foi de 28,8 horas em pacientes com câncer de mama avançado.
Em seis homens saudáveis que receberam dose única oral de [14C]Palbociclibe (substância ativa), uma mediana de 91,6% da dose radioativa total administrada foi recuperada em 15 dias; as fezes (74,1% da dose) foram a principal via de excreção, com 17,5% da dose recuperada na urina. A excreção de Palbociclibe (substância ativa) inalterado nas fezes e na urina foi 2,3% e 6,9% da dose administrada, respectivamente.
Idade, sexo e peso corporal
Com base na análise farmacocinética populacional em 183 pacientes com câncer (50 homens e 133 mulheres, com idades entre 22 e 89 anos e peso corporal entre 37,9 a 123 kg), o sexo não teve efeito na exposição ao Palbociclibe (substância ativa), e idade e peso corporal não tiveram efeito clinicamente importante na exposição ao Palbociclibe (substância ativa).
População pediátrica
A farmacocinética do Palbociclibe (substância ativa) não foi avaliada em pacientes com idade ?18 anos.
População idosa
Dos 444 pacientes que receberam Palbociclibe (substância ativa) no Estudo 2, 181 pacientes (41%) tinham ?65 anos de idade. Dos 347 pacientes que receberam Palbociclibe (substância ativa) no Estudo 3, 86 pacientes (24,8%) tinham ?65 anos de idade. Nenhuma diferença geral na segurança ou eficácia do Palbociclibe (substância ativa) foi observada entre esses pacientes e pacientes mais jovens.
Insuficiência hepática
Os dados de um estudo farmacocinético em indivíduos com diferentes graus de função hepática indicam que a exposição não ligada com Palbociclibe (substância ativa) (AUCinf não ligado) diminuiu 17% em indivíduos com insuficiência hepática leve (Child-Pugh classe A) e aumentou 34% e 77% em indivíduos com insuficiência hepática moderada (Child-Pugh classe B) e grave (Child-Pugh classe C), respectivamente, em relação a indivíduos com função hepática normal. Pico de Palbociclibe (substância ativa) em exposição não ligada (Cmax não ligada) aumentou 7%, 38% e 72% para insuficiência hepática leve, moderada e grave, respectivamente, em relação a indivíduos com função hepática normal.
Além disso, com base em uma análise farmacocinética populacional que incluiu 183 pacientes com câncer avançado, onde 40 pacientes apresentaram comprometimento hepático leve com base na classificação do Cancer National Institute (NCI) (bilirrubina total ? Limite superior de Normal (ULN) e Aspartato Aminotransferase (AST)> ULN, ou bilirrubina total > 1,0 a 1,5 × ULN e qualquer AST). Comprometimento hepático leve não teve efeito sobre a farmacocinética (PK) do Palbociclibe (substância ativa).
Insuficiência renal
Os dados de um estudo farmacocinético em indivíduos com diferentes graus de função renal indicam que a exposição total ao Palbociclibe (substância ativa) (AUCinf) foi aumentada em 39%, 42% e 31% com insuficiência renal leve (60 mL/min?CrCl<90mL/min), moderada (30 mL/min?CrCl<60 mL/min) e grave (CrCl<30 mL/min), respectivamente, em relação a indivíduos com função renal normal (CrCl ?90 mL/min).
Pico de exposição ao Palbociclibe (substância ativa) (Cmax) aumentou 17%, 12% e 15% para insuficiência renal leve, moderada e grave, respectivamente, em relação aos indivíduos com função renal normal.
Além disso, com base em uma análise farmacocinética populacional que incluiu 183 pacientes com câncer avançado, em que 73 pacientes apresentaram insuficiência renal leve e 29 pacientes apresentaram insuficiência renal moderada, insuficiência renal leve e moderada não tiveram efeito sobre a PK do Palbociclibe (substância ativa). A farmacocinética do Palbociclibe (substância ativa) não foi estudada em pacientes que necessitem de hemodiálise.
Raça asiática
Em um estudo farmacocinético em voluntários sadios, os valores AUCinf e Cmáx do Palbociclibe (substância ativa) foram 30% e 35% maiores, respectivamente, em indivíduos japoneses, quando comparados com indivíduos não asiáticos após uma dose única oral. No entanto, essa descoberta não foi reproduzida consistentemente em estudos subsequentes em pacientes japoneses ou asiáticos com câncer de mama após administração múltipla. Com base em uma análise dos dados cumulativos de farmacocinética, segurança e eficácia em populações asiáticas e não asiáticas, não é considerado necessário um ajuste da dose com base na raça asiática.
Eletrofisiologia cardíaca
O efeito de Palbociclibe (substância ativa) no intervalo QT corrigido de frequência cardíaca (QTc) foi avaliado usando eletrocardiogramas (ECGs) controlado por tempo, avaliando a mudança na avaliação inicial e nos dados farmacocinéticos correspondentes em 77 pacientes com câncer de mama. O Palbociclibe (substância ativa) não prolongou o QTc em qualquer grau clinicamente relevante na dose diária recomendada de 125 mg (Esquema 3/1).
Dados de segurança pré-clínicos
Os achados primários de órgãos-alvo após dosagem única e/ou repetida incluíram efeitos sobre órgãos hematolinfopoiéticos e reprodutores masculinos em ratos e cachorros, e efeitos em ossos e incisivos com crescimento ativo somente em ratos. Estas toxicidades sistêmicas foram observadas, de modo geral, em exposições clinicamente relevantes com base na AUC. Estabeleceu-se reversão parcial, ou total, dos efeitos sobre o sistema hematolinfopoiético, sistemas reprodutores masculinos e dentes incisivos, ao passo que o efeito em ossos não foi revertido após um período sem dosagem de 12 semanas. Além disso, efeitos cardiovasculares (prolongamento de QTc, diminuição da frequência cardíaca e aumento no intervalo RR e na pressão arterial sistólica) foram identificados em cachorros por telêmetro em ?4 vezes a exposição clínica em humanos, com base na Cmáx.
Carcinogenicidade
Estudos de carcinogenicidade não foram conduzidos com Palbociclibe (substância ativa).
Genotoxicidade
O Palbociclibe (substância ativa) não foi mutagênico em um ensaio de mutação reversa bacteriana (Ames) e não induziu aberrações cromossômicas estruturais no ensaio de aberração cromossômica de linfócito humano in vitro.
O Palbociclibe (substância ativa) induziu micronúcleos via mecanismo aneugênico em células de ovário de hamster chinês in vitro e na medula óssea de ratos machos em doses de ?100 mg/kg/dia. O nível de efeito não observado para a aneugenicidade foi de aproximadamente sete vezes a exposição clínica em humanos, baseado na AUC.
Comprometimento de fertilidade
O Palbociclibe (substância ativa) não afetou o acasalamento ou fertilidade de ratas fêmeas testadas com até 300 mg/kg/dia (aproximadamente três vezes a exposição clínica em humanos baseada na AUC) e nenhum efeito adverso foi observado em tecidos reprodutivos femininos nos estudos de toxicidade de dose repetida de até 300 mg/kg/dia em ratos e 3 mg/kg/dia em cachorros (aproximadamente cinco e três vezes a exposição clínica em humanos na AUC, respectivamente).
Considera-se que Palbociclibe (substância ativa) tenha o potencial de prejudicar a função reprodutora e a fertilidade em homens com base nos achados não clínicos observados em ratos e cachorros. Os achados relacionados ao Palbociclibe (substância ativa) nos testículos, epidídimos, próstata e vesícula seminal incluíram redução no peso dos órgãos, atrofia ou degeneração, hipospermia, debris celulares intratubulares, menor motilidade e densidade de espermatozoides e secreção reduzida. Esses achados foram observados em ratos e/ou cachorros em exposições ?9 vezes a exposição clínica em humanos, ou em comparações subterapêuticas, com base na AUC. A reversibilidade parcial dos efeitos nos órgãos reprodutivos masculinos foi observada em ratos e cachorros após um período de quatro e doze semanas sem administração, respectivamente. Apesar destas descobertas sobre o órgão reprodutor masculino, não houve nenhum efeito sobre o acasalamento ou sobre a fertilidade de ratos machos em níveis de exposição projetada de 13 vezes a exposição clínica em humanos, com base na AUC.
Toxicidade no desenvolvimento
O Palbociclibe (substância ativa) foi fetotóxico em fêmeas grávidas. Um aumento na incidência de uma variação esquelética (aumento da incidência de uma costela presente na sétima vértebra cervical) foi observado em ratas que receberam ?100 mg/kg/dia. Observou-se em ratas peso corpóreo fetal reduzido em uma dose tóxica para a mãe, de 300 mg/kg/dia (três vezes a exposição clínica em humanos, com base na AUC), e foi observado em coelhas um aumento na incidência de variações esqueléticas, incluindo pequenas falanges nos membros dianteiros, em uma dose tóxica para a mãe, de 20 mg/kg/dia (quatro vezes a exposição clínica em humanos, com base na AUC).
A exposição fetal real e a transferência de placenta cruzada não foram examinadas.
Cuidados de Armazenamento do Ibrance
Ibrance deve ser conservado em temperatura ambiente (entre 15 e 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características do produto
Dizeres Legais do Ibrance
MS - 1.0216.0257
Farmacêutica Responsável:
Carolina C. S. Rizoli
CRF-SP nº 27071
Registrado por:
Laboratórios Pfizer Ltda.
Rodovia Presidente Castelo Branco, n° 32.501, Km 32,5
CEP 06696-000 - Itapevi - SP
CNPJ nº 46.070.868/0036-99
Fabricado por:
Pfizer Manufacturing Deutschland GmbH – Betriebsstätte Freiburg
Freiburg - Alemanha
Venda sob prescrição médica.
informações complementares
| Fabricante |
| LABORATORIOS WYETH-WHITEHALL |
| Princípio ativo |
| Palbociclibe |
| Categoria do medicamento |
| Medicamentos Especiais |