para o que é indicado e para que serve?
Para que serve Jakavi é um medicamento usado para tratar pacientes adultos com mielofibrose de risco intermediário ou alto, um tipo raro de câncer do sangue com vários sintomas incômodos como febre, sudorese noturna, dor nos ossos e perda de peso.Continue lendo...
ofertas de
Jakavi - 20 Mg 60 Comprim...
ofertas de Jakavi - 20 Mg 60 Comprim...
R$ 28.114,56
R$ 28.220,00
R$ 28.747,00
R$ 28.747,00
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Jakavi é um medicamento usado para tratar pacientes adultos com mielofibrose de risco intermediário ou alto, um tipo raro de câncer do sangue com vários sintomas incômodos como febre, sudorese noturna, dor nos ossos e perda de peso.
O aumento do baço é uma das características da mielofibrose.
Como o Javaki funciona?
Jakavi contém o princípio ativo ruxolitinibe.
A mielofibrose é um distúrbio da medula óssea, no qual a medula é substituída por tecido fibroso. A medula alterada não consegue mais produzir uma quantidade suficiente de células sanguíneas e isso resulta no aumento significativo do baço.
Jakavi pode reduzir o tamanho do baço em pacientes com diferentes tipos de mielofibrose ao bloquear de forma seletiva as enzimas denominadas Janus Quinases Associadas (JAK1 e JAK2), aliviando assim os sintomas e reduzindo o risco de complicações sanguíneas ou vasculares possivelmente graves.
Consulte seu médico em caso de dúvidas sobre como Jakavi funciona ou por que este medicamento foi prescrito para você.
Contraindicação
- Se você for alérgico (hipersensível) ao ruxolitinibe ou a qualquer outro componente de Jakavi listado no início da bula. Consulte seu médico se você acredita que pode ser alérgico.
Se isso aplicar a você, informe ao seu médico antes de iniciar o tratamento com Jakavi.
Como usar
Siga cuidadosamente as instruções do seu médico. Não tome mais Jakavi além do que foi prescrito pelo seu médico.
Como tomar Jakavi
Os comprimidos de Jakavi devem ser ingeridos por via oral com ou sem alimentos.
Os comprimidos devem ser ingeridos inteiros com um copo de água.
Quanto tomar de Jakavi
Seu médico lhe dirá exatamente quantos comprimidos de Jakavi você deve tomar.
Seu médico irá verificar suas células sanguíneas e a condição do seu fígado e rins para determinar e manter a dose de Jakavi adequada para você.
O médico também precisa saber se você está se tratando com certos medicamentos, certifique-se de informá-lo sobre outros medicamentos que você esteja tomando.
Se você apresentar determinados efeitos colaterais decorrentes do uso de Jakavi (por exemplo, distúrbios do sangue) talvez seja preciso que seu médico altere a quantidade de Jakavi que você deve tomar ou instrua-o a parar de tomar Jakavi durante algum tempo.
Não interrompa o tratamento com Jakavi, a menos que o seu médico oriente-lhe de outra forma.
Quando tomar Jakavi
Você deve tomar Jakavi duas vezes ao dia, todos os dias, aproximadamente no mesmo horário.
É importante tomar Jakavi aproximadamente no mesmo horário todos os dias para que haja uma quantidade regular na corrente sanguínea.
Se você faz diálise, deve tomar uma dose única de Jakavi antes e outra dose única depois do término da diálise e somente no dia da diálise. Seu médico irá informá-lo qual a dose única que você deve tomar antes e depois da diálise.
Por quanto tempo tomar Jakavi
Você deve continuar tomando Jakavi pelo tempo que seu médico determinar.
Este é um tratamento a longo prazo. Seu médico irá monitorar regularmente a sua condição para garantir que o tratamento esteja surtindo o efeito esperado.
Converse com seu médico ou farmacêutico em caso de dúvidas sobre quanto tempo tomar Jakavi.
Se você parar de tomar Jakavi
Se você interromper o tratamento com Jakavi, os sintomas relacionados à mielofibrose podem reaparecer. Portanto, você não deve parar de tomar Jakavi sem o consentimento do seu médico.
Consulte seu médico ou farmacêutico em caso de dúvidas sobre o uso deste produto.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que devo fazer quando eu me esquecer de usar este medicamento?
Não tome uma dose dupla de Jakavi para compensar uma dose esquecida.
Se você se esquecer de tomar Jakavi, simplesmente tome a próxima dose no horário planejado.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Siga cuidadosamente todas as instruções do médico. Elas podem ser diferentes das informações gerais contidas nesta bula.
Antes de iniciar o tratamento com Jakavi
Informe ao seu médico se você:
- Tem alguma infecção;
- Tem algum problema nos rins;
- Tem ou já teve problemas no fígado;
- Está tomando outros medicamentos;
- Já teve tuberculose;
- Já teve câncer de pele;
- Já teve hepatite B viral.
Durante o tratamento com Jakavi
Esse medicamento pode provocar uma diminuição temporária no número de células sanguíneas no seu corpo. Isso pode aumentar o risco de desenvolver infecções graves ou sangramentos.
Informe ao seu médico imediatamente se você:
- Apresentar hematomas e/ou sangramentos inesperados, cansaço incomum, falta de ar com exercícios ou em repouso, palidez ou infecções frequentes (sinais de distúrbios do sangue);
- Apresentar sintomas de infecções ou desenvolver erupções cutâneas dolorosas com bolhas (sinais de herpes zoster);
- Apresentar tosse crônica com escarro com sangue, febre, sudorese noturna e perda de peso (estes são sinais de tuberculose);
- Apresentar qualquer um dos seguintes sintomas ou se alguém próximo a você perceber que você tem algum destes sintomas: confusão ou dificuldade de raciocínio, perda de equilíbrio ou dificuldade ao andar, falta de jeito, dificuldade ao falar, diminuição da força ou ter fraqueza em um lado do seu corpo, visão turva e/ou perda da visão (estes são sinais de leucoencefalopatia multifocal progressiva);
- Notar alterações na pele. Isso pode exigir um acompanhamento, uma vez que certos tipos de câncer de pele (não melanoma) foram relatados.
Monitoramento durante o tratamento com Jakavi
Antes de começar seu tratamento com Jakavi, o médico realizará exames de sangue para determinar a sua dose inicial.
Seu médico irá verificar cuidadosamente se você apresenta quaisquer sinais ou sintomas de infecção antes de iniciar e durante seu tratamento com Jakavi.
Alguns exames de sangue serão realizados durante o tratamento com Jakavi para monitorar a quantidade de células sanguíneas no seu organismo (glóbulos brancos e vermelhos, plaquetas) para observar o modo como você responde ao tratamento.
Se Jakavi estiver causando um efeito indesejado sobre estas células talvez seja preciso que seu médico ajuste a dose ou interrompa o tratamento com Jakavi.
Seu médico pode também regularmente verificar o nível de lipídios (gordura) no seu sangue.
Interrupção do tratamento
Após a interrupção do tratamento com Jakavi, os pacientes podem experimentar um retorno dos sintomas da mielofibrose, tais como:
- Fadiga;
- Dor óssea;
- Febre;
- Prurido;
- Suores noturnos;
- Esplenomegalia;
- Perda de peso.
O médico pode reduzir gradualmente a dose diária de Jakavi, antes de interromper completamente o tratamento.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como ocorre com todos os medicamentos, os pacientes que tomam Jakavi podem apresentar efeitos colaterais, embora nem todas as pessoas desenvolvam tais efeitos.
A maioria dos efeitos colaterais é leve a moderado e geralmente desaparece depois de alguns dias a algumas semanas de tratamento.
Reações adversas muito comuns (ocorre em mais de 10% dos pacientes que utilizam este medicamento)
- Infecção do trato urinário;
- Cansaço, fadiga, pele pálida (possíveis sintomas de anemia, que são causados pelo baixo nível de células vermelhas no sangue), infecções frequentes, febre, calafrios, dor de garganta ou úlceras na boca devido à infecções (possíveis sintomas de neutropenia, que são causados pelo baixo nível de células brancas no sangue), sangramento ou hematomas espontâneos (possíveis sintomas de trombocitopenia que são causados por baixos níveis de plaquetas);
- Nível alto de colesterol (hipercolesterolemia);
- Tontura;
- Dor de cabeça;
- Resultados alterados dos testes de função hepática;
- Hematomas;
- Ganho de peso;
- Sangramento, sangramento nasal, sangramento pós-procedimento e sangue na urina;
- Alguns testes que são realizados em relação à função hepática (aumento da alanina aminotransferase e aspartato aminotransferase) podem ser afetados.
Reações adversas comuns (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento)
- Herpes zoster;
- Excesso de gases nos intestinos (flatulência);
- Qualquer sinal de sangramento intracraniano, tais como alteração do nível de consciência, dor de cabeça persistente, dormência, formigueiro, fraqueza ou paralisia;
- Qualquer sinal de sangramento gastrointestinal, tais como fezes de cor negra ou ensanguentadas ou vômitos com sangue.
Reações adversas incomuns (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento)
- - Tosse crônica com escarro tingido de sangue, febre, suores noturnos e perda de peso (sintomas de tuberculose).
Após a interrupção do tratamento com Jakavi, os pacientes podem experimentar um retorno dos sintomas da mielofibrose, tais como fadiga, dor óssea, febre, prurido, suores noturnos, esplenomegalia e perda de peso.
O médico pode reduzir gradualmente a dose diária de Jakavi, antes de interromper completamente o tratamento.
Alguns testes que são realizados em relação à função hepática (aumento da alanina aminotransferase e aspartato aminotransferase) podem ser afetados.
Informe ao seu médico se algum desses efeitos lhe afetar gravemente.
Informe seu médico ou farmacêutico se você observar algum efeito colateral não mencionado nesta bula.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
População Especial
Pacientes idosos (65 anos de idade ou mais)
Jakavi pode ser usado por pacientes de 65 anos ou mais sem necessidade de ajuste da dose.
Crianças e adolescentes (menores de 18 anos de idade)
Jakavi não deve ser utilizado por crianças ou adolescentes.
Gravidez e Lactação
Consulte seu médico ou farmacêutico antes de tomar qualquer medicamento.
Seu médico irá aconselhá-la a tomar as medidas apropriadas para evitar gravidez durante o tratamento com Jakavi.
O uso de Jakavi não é recomendado durante a gravidez, a menos que claramente necessário.
Se você estiver grávida ou acreditar que possa estar grávida é importante informar seu médico, que discutirá com você se é possível tomar Jakavi durante sua gravidez.
Você não deve amamentar seu filho enquanto estiver tomando Jakavi. Não se sabe se Jakavi passa para o leite materno.
Efeitos na habilidade de dirigir veículos e/ou operar máquinas
Se você apresentou tonturas durante o tratamento com Jakavi não deve conduzir veículos ou operar máquinas, pois sua habilidade e atenção podem estar prejudicadas.
Composição
Cada comprimido de Jakavi 5 mg contém
- 6,60 mg de fosfato de ruxolitinibe (equivalente a 5 mg de ruxolitinibe).
Cada comprimido de Jakavi 15 mg contém
-
- 19,80 mg de fosfato de ruxolitinibe (equivalente a 15 mg de ruxolitinibe).
Cada comprimido de Jakavi 20 mg contém
- 26,40 mg de fosfato de ruxolitinibe (equivalente a 20 mg de ruxolitinibe).
Excipientes: lactose monoidratada, celulose microcristalina, amidoglicolato de sódio, hiprolose, povidona, dióxido de silício, estearato de magnésio.
Superdosagem
Entre imediatamente em contato com seu médico, enfermeiro ou farmacêutico se você tomar acidentalmente mais Jakavi do que o médico prescreveu.
Sintomas que podem caracterizar uma superdose
- Sangramento incomum;
- Tonturas;
- Dor de cabeça;
- Cansaço;
- Febre;
- Dor de garganta;
- Calafrios;
- Tosse;
- Outros sinais de infecção: leucopenia, anemia e trombocitopenia.
Orientações gerais quanto a medidas preventivas
Ruxolitinibe foi administrado com segurança em doses até 100 mg, uma vez ao dia, durante pelo menos 10 dias consecutivos.
As toxicidades potenciais com overdose de ruxolitinibe são raras, pouco provável que seja de natureza aguda (imediata) e os efeitos são potencialmente retardados como consequência da inibição da JAK1/2. Portanto, como não existem riscos potenciais de overdose com ruxolitinibe de caráter imediato (ou aguda), não há medidas de emergência ou preventivas necessárias ou que possam ser recomendadas como uma orientação geral, além de buscar atenção médica o mais rapidamente possível.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Agentes que podem alterar a concentração plasmática de Ruxolitinibe (substância ativa)
Interações estudadas com outros medicamentos
Fatores de crescimento hematopoiético e terapias citorredutoras
O uso concomitante de terapias citorredutoras ou fatores de crescimento hematopoiético com Ruxolitinibe (substância ativa) não foi estudado. A segurança e eficácia destas coadministrações são desconhecidas.
Interação Alimentícia
Este medicamento pode ser administrado com ou sem alimento.
Ação da Substância
Mielofibrose
Dois estudos randomizados de Fase 3 (COMFORT-I e COMFORT-II) foram conduzidos em pacientes com Mielofibrose (MF) (Mielofibrose Primária (MFP), Mielofibrose Pós-Policitemia Vera (MF-PPV) ou Mielofibrose Pós-Trombocitemia Essencial (MF-PTE)). Nos dois estudos, os pacientes apresentaram esplenomegalia palpável pelo menos 5 cm abaixo da margem costal e categoria de risco intermediário 2 (2 fatores prognósticos) ou alto risco (3 ou mais fatores prognósticos) com base nos Critérios de Consenso do Grupo de Trabalho Internacional (IWG). Os fatores prognósticos que compreendem os critérios do IWG consistem em idade > 65 anos, presença de sintomas constitucionais (perda de peso, febre, sudorese noturna), anemia (hemoglobina < 10 g/dL), leucocitose (história de contagem de leucócitos > 25 x 109/L) e blastos circulantes ? 1%. A dose inicial de Ruxolitinibe (substância ativa) teve como base a contagem de plaquetas. Pacientes com uma contagem de plaquetas entre 100.000 e 200.000/mm3 iniciaram Ruxolitinibe (substância ativa) 15 mg, duas vezes ao dia, e pacientes com uma contagem de plaquetas > 200.000/mm3 iniciaram Ruxolitinibe (substância ativa) 20 mg, duas vezes ao dia.
As doses foram, então, individualizadas com base na tolerabilidade e na eficácia, com doses máximas de 20 mg, duas vezes ao dia, para pacientes com contagens de plaquetas entre 100.000 a ? 125.000/mm3, de 10 mg, duas vezes ao dia, para pacientes com contagens de plaquetas entre 75.000 a ? 100.000/mm3, e de 5 mg, duas vezes ao dia, para pacientes com contagens de plaquetas entre 50.000 a ? 75.000/mm3.
COMFORT-I foi um estudo duplo-cego, randomizado, controlado por placebo em 309 pacientes refratários ou que não eram candidatos para a terapia disponível. Os pacientes receberam doses de Ruxolitinibe (substância ativa) ou placebo correspondente. O objetivo primário de eficácia foi a proporção de indivíduos que atingiram redução ? 35% no volume do baço desde o basal na Semana 24, conforme medição por ressonância magnética (RM) ou tomografia computadorizada (TC).
Os objetivos secundários incluíram a duração da manutenção da redução ? 35% desde o basal no volume do baço, proporção de pacientes que tiveram redução ? 50% na pontuação total de sintomas desde o basal até a Semana 24, conforme medição no diário do Formulário de Avaliação dos Sintomas de Mielofibrose Modificado (FASMM) v2.0, alteração na pontuação total de sintomas desde o basal até a Semana 24, conforme medição no diário do FASMM v2.0 modificado e sobrevida global.
COMFORT-II foi um estudo randomizado e aberto em 219 pacientes. Os pacientes foram randomizados em uma proporção de 2:1 para Ruxolitinibe (substância ativa) versus melhor terapia disponível. A melhor terapia disponível foi escolhida pelo investigador caso a caso. No braço de melhor terapia disponível, 47% dos pacientes receberam hidroxiureia e 16% dos pacientes receberam glicocorticoides. O objetivo primário de eficácia foi a proporção de pacientes que atingiu redução ? 35% no volume do baço desde o basal na Semana 48, conforme medição por RM ou TC.
Um objetivo secundário no COMFORT-II foi a proporção de pacientes que atingiu redução ? 35% no volume do baço medida por RM ou TC desde o basal até a Semana 24. A duração da manutenção de redução ? 35% desde o basal nos pacientes respondedores também foi um objetivo secundário.
No COMFORT-I, os dados demográficos do basal dos pacientes e as características da doença foram semelhantes entre os braços de tratamento. A idade mediana foi de 68 anos, com 61% dos pacientes com mais de 65 anos de idade e 54% sendo homens. Cinquenta por cento (50%) dos pacientes apresentaram mielofibrose primária, 31% apresentaram mielofibrose pós-policitemia e 18% apresentaram mielofibrose pós-trombocitemia essencial. Vinte e um (21%) dos pacientes tiveram transfusões de sangue em até 8 semanas a partir da inclusão no estudo. A contagem mediana de plaquetas foi de 251.000/mm3. Setenta e seis por cento dos pacientes apresentaram a mutação, codificando a substituição V617F presente na proteína JAK. Os pacientes tiveram um comprimento de baço mediano palpável de 16 cm. No basal, 37,4% dos pacientes no braço Ruxolitinibe (substância ativa) apresentaram anemia grau 1, 31,6% grau 2 e 4,5% grau 3, enquanto que no braço de placebo, 35,8% apresentaram grau 1, 35,1% grau 2, 4,6% grau 3 e 0,7% grau 4. Trombocitopenia grau 1 foi encontrada em 12,9% dos pacientes no braço de Ruxolitinibe (substância ativa) e 13,2% no braço de placebo.
No COMFORT-II, os dados demográficos do basal dos pacientes e as características da doença foram semelhantes entre os braços de tratamento. A idade mediana foi de 66 anos, com 52% dos pacientes com mais de 65 anos de idade e 57% sendo homens. Cinquenta e três por cento (53%) dos pacientes apresentaram mielofibrose primária, 31% apresentaram mielofibrose pós-policitemia vera e 16% apresentaram mielofibrose pós-trombocitemia essencial. Dezenove por cento (19%) dos pacientes foram considerados dependentes de transfusão no basal. Os pacientes apresentaram um comprimento mediano de baço palpável de 15 cm.
No basal, 34,2% dos pacientes no braço de Ruxolitinibe (substância ativa) apresentaram anemia de grau 1, 28,8% grau 2, e 7,5% grau 3, enquanto que no braço de BAT 37% apresentaram grau 1, 27,4% grau 2, 13,7% grau 3, e 1,4% grau 4. Trombocitopenia de grau 1 foi encontrada em 8,2% dos pacientes no braço de Ruxolitinibe (substância ativa) e 9,6% no braço de BAT 1. As análises de eficácia do objetivo primário no COMFORT-I e COMFORT-II são apresentadas na Tabela 1 abaixo. Uma proporção significativamente maior de pacientes no grupo de Ruxolitinibe (substância ativa) atingiu redução ? 35% no volume do baço desde o basal nos dois estudos em comparação ao placebo no COMFORT-I e melhor terapia disponível no COMFORT-II.
Tabela 1. Percentual de Pacientes com Redução ? 35% desde o Basal no Volume do Baço na Semana 24 no COMFORT-I e na Semana 48 no COMFORT-II (ITT)
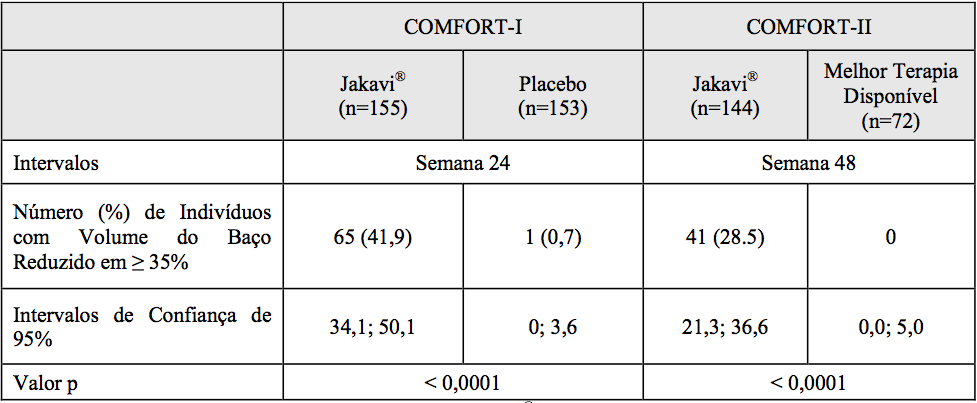
No COMFORT-I, 41,9% dos pacientes no grupo de Ruxolitinibe (substância ativa) atingiram redução ? 35% no volume do baço desde o basal em comparação a 0,7% no grupo de placebo na Semana 24. Uma proporção semelhante de pacientes no grupo de Ruxolitinibe (substância ativa) atingiu redução ? 50% no comprimento do baço palpável.
No COMFORT-II, 28,5% dos pacientes no grupo de Ruxolitinibe (substância ativa) atingiram redução ? 35% no volume do baço desde o basal em comparação a nenhum (0%) no grupo da melhor terapia disponível na Semana 48. Um objetivo secundário foi a proporção de pacientes que atingiu redução ? 35% no volume do baço na Semana 24. Uma proporção significativamente maior de pacientes no grupo de Ruxolitinibe (substância ativa), 46 (31,9%) atingiu redução ? 35% no volume do baço desde o basal em comparação a nenhum (0%) paciente no grupo da melhor terapia disponível (valor p < 0,0001).
Uma proporção significativamente maior de pacientes no grupo de Ruxolitinibe (substância ativa) atingiu redução ? 35% desde o basal no volume do baço independente da presença ou ausência da mutação JAK2V617F ou subtipo da doença (mielofibrose primária, mielofibrose pós-policitemia vera, mielofibrose pós-trombocitemia essencial). A figura 1 apresenta um gráfico em cascata da alteração percentual desde o basal no volume do baço na Semana 24 no COMFORT-I. Entre os 139 pacientes no grupo de Ruxolitinibe (substância ativa) que apresentaram as duas avaliações do volume do baço no basal e na Semana 24, todos exceto dois pacientes tiveram algum nível de redução no volume do baço na Semana 24, com redução mediana de 33%. Entre os 106 pacientes no grupo de placebo que apresentaram avaliações do volume do baço no basal e na Semana 24, houve um aumento mediano de 8,5%.
Figura 1. Gráfico em Cascata da Alteração Percentual Desde o Basal no Volume do Baço na Semana 24 (Casos Observados) COMFORT-I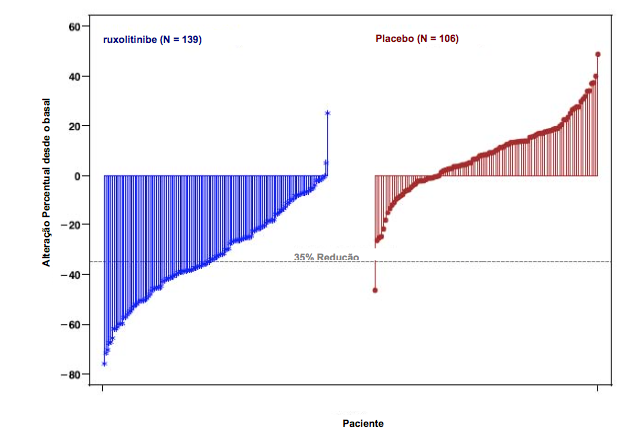
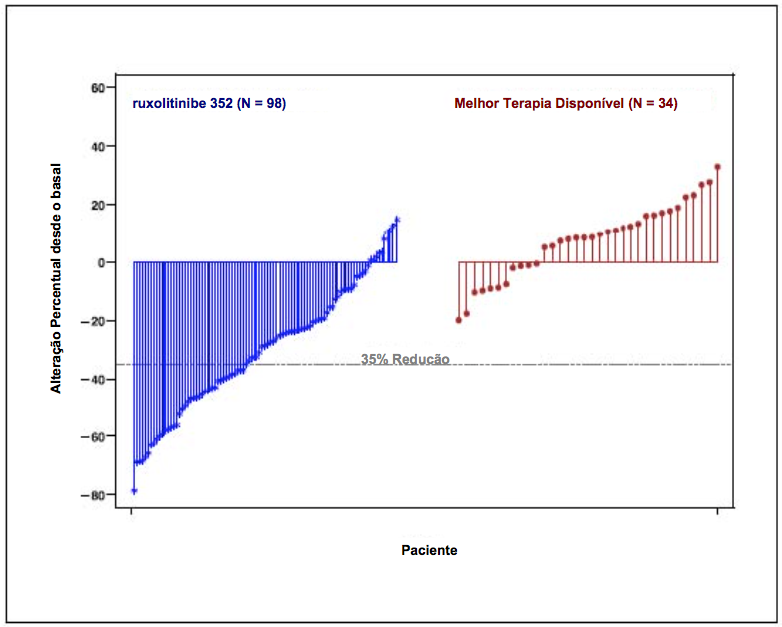
A figura 2 apresenta um gráfico em cascata da alteração percentual desde o basal no volume do baço na Semana 48 no COMFORT-II. Entre os 98 pacientes no grupo de Ruxolitinibe (substância ativa) que apresentaram as duas avaliações do volume do baço no basal e na Semana 48, a redução mediana no volume do baço na Semana 48 foi de 28%. Entre os 34 pacientes no grupo da Melhor Terapia Disponível que apresentaram avaliações do volume do baço no basal e na Semana 48, houve um aumento mediano de 8,5%.
Figura 2. Gráfico em Cascata da Alteração Percentual desde o Basal no Volume do Baço na Semana 48 no COMFORT-II
A probabilidade de duração da 1a redução ? 35% do volume do baço até um aumento de 25% desde o nadir e perda de resposta no COMFORT-I e COMFORT-II é apresentado na Tabela 2 abaixo.
Tabela 2. Análise de Kaplan-Meier da Duração da 1a Redução ? 35% do Volume do Baço Até um Aumento de 25% desde o Nadir e Perda de Resposta em Pacientes Recebendo Ruxolitinibe (substância ativa) (COMFORT-I e II)
| Estatística | Ruxolitinibe (substância ativa)(COMFORT-I) | Ruxolitinibe (substância ativa)(COMFORT-II) |
| Probabilidade de duração > 12 semanas (IC de 95%) | 0,98 (0,89; 1,00) | 0,92 (0,82; 0,97) |
| Probabilidade de duração > 24 semanas (IC de 95%) | 0,89 (0,75; 0,95) | 0,87 (0,76; 0,93) |
| Probabilidade de duração > 36 semanas (IC de 95%) | 0,71 (0,41; 0,88) | 0,77 (0,63; 0,87) |
| Probabilidade de duração > 48 semanas (IC de 95%) | não aplicável | 0,52 (0,18; 0,78) |
Entre os 80 pacientes que apresentaram redução ? 35% em qualquer momento no COMFORT-I e os 69 pacientes no COMFORT-II, a probabilidade de um paciente manter uma resposta com Ruxolitinibe (substância ativa) por pelo menos 24 semanas foi de 89% e 87% no COMFORT-I e COMFORT-II, respectivamente, e a probabilidade de manutenção da resposta por pelo menos 48 semanas foi de 52% no COMFORT-II.
Ruxolitinibe (substância ativa) melhora os sintomas relacionados à mielofibrose e qualidade de vida (QOL) em pacientes com MFP, MF-PPV e MF-PTE. No COMFORT-I, os sintomas de MF foram capturados utilizando-se o diário do Formulário de Avaliação dos Sintomas de Mielofibrose Modificado (FASMM) v2.0 como um diário eletrônico, o qual os indivíduos preenchiam diariamente. A alteração desde o basal na pontuação total na Semana 24 foi um objetivo secundário neste estudo. Uma proporção significativamente maior de indivíduos no grupo de Ruxolitinibe (substância ativa) atingiu melhora ? 50% desde o basal na pontuação total dos sintomas na semana 24 comparado ao grupo placebo (45,9% e 5,3%, respectivamente, p < 0,0001 usando o teste do Qui-quadrado).
Uma melhora na qualidade de vida global foi medida pelo EORTC QLQ-C30 no COMFORT-I e no COMFORT-II. COMFORT-I comparou Ruxolitinibe (substância ativa) com placebo por 24 semanas e COMFORT-II comparou Ruxolitinibe (substância ativa) com a melhor terapia disponível por 48 semanas. No basal, para os dois estudos, as pontuações da subescala individual de EORTC QLQ-C30 para os grupos de Ruxolitinibe (substância ativa) e comparador foram similares. Na Semana 24, no COMFORT-I, o grupo de Ruxolitinibe (substância ativa) demonstrou uma melhora significativa da saúde global/qualidade de vida do EORTC QLQ-C30 comparado ao grupo de placebo (alteração média de +12,3 e - 3,4 para Ruxolitinibe (substância ativa) e placebo, respectivamente, p < 0,0001). Na semana 24 e na semana 48, o grupo de Ruxolitinibe (substância ativa) no COMFORT-II apresentou uma tendência em direção a uma melhora maior da saúde global/qualidade de vida comparada à melhor terapia disponível, um objetivo exploratório, consistente com os achados do COMFORT-II.
No COMFORT-I, após acompanhamento médio de 34,3 meses, a taxa de mortes em pacientes randomizados no braço Ruxolitinibe (substância ativa) foi de 27,1% (42 de 155 pacientes) versus 35,1% (54 de 154) dos pacientes randomizados com placebo. Houve uma redução de 31,3% no risco de morte no braço Ruxolitinibe (substância ativa) quando comparado ao placebo (HR 0,687; IC 95% 0,459-1,029; p = 0,0668).
No COMFORT-I, após acompanhamento médio de 61,7 meses, a taxa de mortes em pacientes randomizados no braço Ruxolitinibe (substância ativa) foi de 44,5% (69 de 155 pacientes) versus 53,2% (82 de 154) dos pacientes randomizados com placebo. Houve uma redução de 31% no risco de morte no braço Ruxolitinibe (substância ativa) quando comparado ao placebo (HR 0,69; IC 95% 0,50-0,96; p = 0,025).
No COMFORT-II, após acompanhamento médio de 34,7 meses, a taxa de mortes em pacientes randomizados com Ruxolitinibe (substância ativa) foi de 19,9% (29 de 146 pacientes) versus 30,1% (22 de 73 pacientes) em pacientes randomizados com melhor terapia disponível (MTD). Houve uma redução no risco de morte de 52% no braço Ruxolitinibe (substância ativa) comparado ao braço MTD (HR 0,48; IC 95% 0,28-0,85; p = 0,009).
No COMFORT-II, após acompanhamento médio de 55,9 meses, a taxa de mortes em pacientes randomizados com Ruxolitinibe (substância ativa) foi de 40,4% (59 de 146 pacientes) versus 47,9% (35 de 73 pacientes) em pacientes randomizados com melhor terapia disponível (MTD). Houve uma redução no risco de morte de 33% no braço Ruxolitinibe (substância ativa) comparado ao braço MTD (HR 0,67; IC 95% 0,44-1,02; p = 0,062).
Policitemia vera
Um estudo de fase 2, multicêntrico, aberto, randomizado, não controlado e com regime de dose variável foi conduzido para estabelecer a dose de 10 mg de Ruxolitinibe (substância ativa) duas vezes ao dia como uma dose ativa, segura e bem tolerada em pacientes com PV avançada refratários à hidroxiureia ou para quem o tratamento com hidroxiureia estava contraindicado. O estudo consistiu de grupos 1 e 2, que recrutaram 34 pacientes com PV.8 Um estudo (RESPONSE) randomizado, aberto, ativo-controlado, de fase 3,7,9 foi conduzido com 222 pacientes com policitemia vera que eram resistentes ou intolerantes à hidroxiureia. Cento e dez pacientes foram randomizados para o braço de Ruxolitinibe (substância ativa) e 112 pacientes para o braço BAT (Best Available Therapy – Melhor Terapia disponível). A dose inicial de Ruxolitinibe (substância ativa) foi de 10 mg duas vezes ao dia. As doses foram então ajustadas individualmente aos pacientes com base na tolerabilidade e eficácia, com a dose máxima de 25 mg duas vezes ao dia. BAT foi selecionada pelo investigador, paciente por paciente e incluída hidroxiureia (59,5%), interferona/interferona peguilada (1,7%), anagrelida (7,2%), pipobromana (1,8%) e observação (15,3%).
Dados demográficos do basal e as características da doença foram comparáveis entre os dois braços de tratamento. A idade média foi de 60 anos (faixa de 33 a 90 anos). Pacientes no braço de Ruxolitinibe (substância ativa) apresentaram diagnóstico PV por uma média de 8,2 anos e tinham recebido previamente hidroxiureia por uma média de aproximadamente de 3 anos. A maioria dos pacientes (> 80%) tinha recebido pelo menos duas flebotomias nas últimas 24 semanas antes da triagem.
O desfecho primário composto foi a proporção de pacientes que atingiram tanto a ausência de elegibilidade de flebotomia (controle HCT) e ? 35% de redução do volume do baço em relação ao basal na semana 32. A elegibilidade de flebotomia foi definida como HCT > 45% confirmado que é, pelo menos, 3 pontos percentuais maior do que o HCT obtido no basal ou um HCT > 48% confirmado, o que for menor. Desfechos secundários principais incluíram a proporção de pacientes que atingiram o desfecho primário e que permaneceram livres de progressão na semana 48, e a proporção de pacientes que atingiram remissão hematológica completa na semana 32.
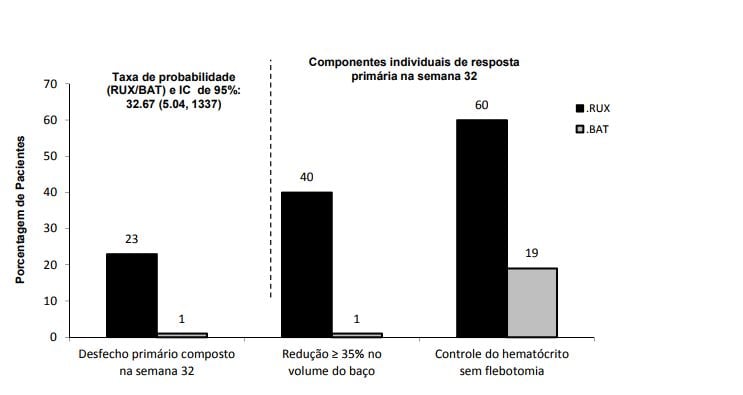
O estudo cumpriu o seu objetivo principal e uma maior proporção de pacientes no grupo Ruxolitinibe (substância ativa) alcançou o desfecho primário composto e cada um dos seus componentes individuais. Significativamente, mais pacientes com Ruxolitinibe (substância ativa) (23%) em comparação com BAT (0,9%) obtiveram uma resposta primária (p < 0,0001). O controle do hematócrito foi conseguido em 60% dos pacientes no braço Ruxolitinibe (substância ativa) em comparação com 18,75% no braço BAT, e redução de ? 35% do volume do baço foi obtido em 40% dos pacientes no braço Ruxolitinibe (substância ativa) em comparação com 0,9% no braço BAT (figura 3).
Ambos os desfechos secundários foram atingidos: a proporção de pacientes que atingiram uma remissão hematológica completa foi de 23,6% com Ruxolitinibe (substância ativa) em comparação a 8,0% com BAT (p = 0,0013), e a proporção de pacientes que atingiram uma resposta primária duradoura na semana 48 foi de 20% com Ruxolitinibe (substância ativa) e de 0,9% com BAT (p < 0,0001).
Figura 3. Pacientes que atingiram o desfecho primário e componentes do desfecho primário na semana 32
Os sintomas foram avaliados usando a pontuação MPN-SAF escore total de sintomas (TSS) do diário eletrônico do paciente que consiste de 14 questões. Na semana 32, 49% e 64% dos pacientes tratados com Ruxolitinibe (substância ativa) conseguiram uma redução ? 50% no TSS-14 e TSS-5, respectivamente, em comparação com apenas 5% e 11% dos pacientes em BAT.
A percepção de benefício do tratamento foi medida pelo questionário Impressão Global de Mudança do Paciente (PGIC). Os 66% dos pacientes tratados com Ruxolitinibe (substância ativa) em comparação com 19% em BAT, relataram uma melhora tão cedo quanto 4 semanas após o início do tratamento. A melhora na percepção de benefício do tratamento também foi maior em pacientes tratados com Ruxolitinibe (substância ativa) na semana 32 (78% versus 33%).
Análises adicionais do estudo RESPONSE para verificar a durabilidade da resposta, foram conduzidas na semana 80 apenas no braço Ruxolitinibe (substância ativa). Neste braço, 83% dos pacientes ainda estavam em tratamento no momento dos dados de corte da semana 80. Dos pacientes que atingiram uma resposta primária na semana 32, 80% mantiveram uma resposta por pelo menos 48 semanas após a resposta inicial. Para os pacientes que atingiram cada um dos componentes do desfecho primário, todos mantiveram a resposta do baço, e a probabilidade de manutenção de controle de hematócrito por pelo menos 80 semanas da resposta inicial foi de 89%. Os 69% dos pacientes que atingiram remissão hematológica completa na semana 32, mantiveram esta resposta por pelo menos 48 semanas.
Um segundo estudo randomizado, aberto, controlado-ativo de fase IIIb (RESPONSE 2)9 , foi conduzido em 149 pacientes com policitemia vera que foram resistentes ou intolerantes à hidroxiureia, mas sem esplenomegalia palpável. Setenta e quatro pacientes foram randomizados para o braço Ruxolitinibe (substância ativa) e 75 pacientes para o braço BAT. A dose inicial e ajustes da dose de Ruxolitinibe (substância ativa) e o BAT selecionado pelo investigador foram semelhantes ao estudo RESPONSE. As características demográficas basais e da doença foram comparadas entre os dois braços de tratamento e foram semelhantes a população de pacientes do estudo RESPONSE. O desfecho primário foi a proporção de pacientes que atingiram o controle de HCT (ausência de elegibilidade de flebotomia) na semana 28. O desfecho chave secundário foi a proporção de pacientes que atingiram a remissão hematológica completa na semana 28.
O estudo RESPONSE-2 cumpriu o seu objetivo primário com uma maior proporção de pacientes no braço Ruxolitinibe (substância ativa) (62,2%) comparado ao braço BAT (18,7%), atingindo seu desfecho primário (p<0,0001). O desfecho chave secundário também foi cumprido com, significativamente, mais pacientes atingindo uma remissão hematológica completa no braço Ruxolitinibe (substância ativa) (23,0%) comparado ao braço BAT (5,3%; p=0,0019). Na semana 28, a proporção de pacientes atingindo uma redução de ? 50% na carga de sintomas como mensurado pela pontuação total de sintomas MPN-SAF foi de 45,3% no braço Ruxolitinibe (substância ativa) e 22,7% no braço BAT.
Características Farmacológicas
Grupo farmacoterapêutico: Agentes antineoplásicos, Inibidor de proteina-quinase.
Código ATC proposto: LO1XE-18.
Propriedades farmacodinâmicas
O Ruxolitinibe (substância ativa) inibe a fosforilação de STAT3 induzida por citocina no sangue total de indivíduos sadios e pacientes com MF. O Ruxolitinibe (substância ativa) resultou na inibição máxima da fosforilação de STAT3 2 horas após a dosagem, e retornou praticamente para o valor basal em 8 horas, tanto em indivíduos sadios quanto em pacientes com mielofibrose, não indicando nenhum acúmulo de metabólitos originais ou ativos.
Elevações do basal nos marcadores inflamatórios associados à sintomas constitucionais como TNF?, IL-6, e CRP em indivíduos com MF haviam diminuído após o tratamento com Ruxolitinibe (substância ativa). Pacientes com mielofibrose não se tornaram refratários aos efeitos farmacodinâmicos do tratamento com Ruxolitinibe (substância ativa) com o passar do tempo.
Em um estudo de QT completo em indivíduos sadios não havia nenhuma indicação quanto ao efeito prolongador do QT/QTc do Ruxolitinibe (substância ativa) em doses únicas até uma dose supraterapêutica de 200 mg, indicando que o Ruxolitinibe (substância ativa) não tem nenhum efeito na repolarização cardíaca.
Propriedades farmacocinéticas
Populações especiais
Dados de segurança pré-clínico
O Ruxolitinibe (substância ativa) foi avaliado em estudos de segurança farmacológica, toxicidade de doses repetidas, genotoxicidade, toxicidade reprodutiva e um estudo de carcinogenicidade. Órgãos alvo associados à ação farmacológica do Ruxolitinibe (substância ativa) em estudos de dose repetida incluem a medula óssea, sangue periférico e tecidos linfoides. Infecções geralmente associadas à imunossupressão foram observadas em cães. Reduções adversas na pressão arterial, juntamente com aumentos na frequência cardíaca, foram observadas em um estudo de telemetria em cães, e uma redução adversa no volume minuto foi observada em um estudo respiratório em ratos. As margens (baseadas na Cmáx não ligada) no nível não adverso em estudos com cães e ratos foram 15,7 vezes e 10,4 vezes maiores, respectivamente, do que a dose diária máxima recomendada para humanos de 25 mg duas vezes ao dia. Não foram observados efeitos em uma avaliação dos efeitos neurofarmacológicos do Ruxolitinibe (substância ativa).
O Ruxolitinibe (substância ativa) não foi teratogênico, mas foi associado à aumentos na perda pós-implantação e reduções nos pesos fetais. Não foram observados efeitos sobre a fertilidade. Em um estudo de desenvolvimento pré e pós- natal, não houve achados adversos quanto a índices de fertilidade e sobrevida materna e embriofetal, crescimento e parâmetros de desenvolvimento. O Ruxolitinibe (substância ativa) não foi mutagênico ou clastogênico. O Ruxolitinibe (substância ativa) não foi carcinogênico no modelo de camundongo transgênico Tg.rasH2 e nem em um estudo de 2 anos em ratos.
Cuidados de Armazenamento
Você deve armazenar este medicamento em temperatura ambiente (entre 15 e 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico
Jakavi é fornecido na forma de comprimidos.
Os comprimidos de 5 mg são redondos e brancos, os de 15 mg são ovais e brancos e os de 20 mg são alongados e brancos.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Mensagens de Alerta
Todo medicamento deve ser mantido fora do alcance das crianças.
Aviso: este medicamento contém lactose.
Pacientes com problemas hereditários de intolerância à galactose, deficiência de lactase de Lapp ou má absorção de glucose-galactose não devem utilizar este medicamento.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista. Informe imediatamente seu médico em caso de suspeita de gravidez.
Dizeres Legais
MS – 1.0068.1121
Farm. Resp.: Flavia Regina Pegorer - CRF-SP 18.150
Importado por:
Novartis Biociências S.A.
Av. Prof. Vicente Rao, 90 - São Paulo - SP
CNPJ: 56.994.502/0001-30
Indústria Brasileira
Fabricado por: Novartis Pharma Stein AG, Stein – Suíça
Venda sob prescrição médica.
Esta bula foi aprovada pela Anvisa em 25/08/2016.
informações complementares
| Fabricante |
| NOVARTIS |
| Princípio ativo |
| Fosfato De Ruxolitinibe |
| Categoria do medicamento |
| Medicamentos Especiais |