para o que é indicado e para que serve?
Para que serve Lemtrada é indicado para o tratamento de pacientes com formas reincidentes de esclerose múltipla (EM) para diminuir ou reverter o acúmulo de incapacidade física e reduzir a frequência de manifestações clínicas.Continue lendo...
ofertas de Lemtrada - 10 Mg/Ml Solução Diluente Infus Frasco-Ampola
R$ 47.410,23
Ir para a lojaR$ 48.876,53
Ir para a lojaR$ 53.898,70
Ir para a lojaR$ 53.898,70
Ir para a lojaATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Lemtrada é indicado para o tratamento de pacientes com formas reincidentes de esclerose múltipla (EM) para diminuir ou reverter o acúmulo de incapacidade física e reduzir a frequência de manifestações clínicas.
Como Lemtrada funciona?
O modo pelo qual Lemtrada ajuda na esclerose múltipla não é conhecido, mas pode envolver um efeito sobre o sistema imunológico, através da diminuição de linfócitos (um tipo de célula branca do sangue). O nível mais baixo de linfócitos foi observado um mês depois do tratamento. Lemtrada age no seu sistemaimunológico para que ele não ataque tanto o seu sistema nervoso.
Contraindicação
Você não deve receber Lemtrada se você tiver reação alérgica conhecida grave ou imediata ao alentuzumabe ou a algum dos ingredientes inativos do medicamento ou ainda se você estiver infectado pelo vírus da Imunodeficiência Humana Adquirida (HIV).
Como usar
Você receberá Lemtrada através de uma injeção na veia, por uma linha de infusão. A administração de uma dose completa em cada dia demora cerca de quatro horas. Você receberá Lemtrada em dois ciclos de tratamento. Em geral, você receberá Lemtrada durante cinco dias para o primeiro ciclo de tratamento e, então, durante três dias aproximadamente um ano depois (segundo ciclo de tratamento).
O seu médico irá retirar 1,2 mL de Lemtrada do frasco-ampola e injetar em 100 mL de solução de cloreto de sódio a 0,9% ou glicose a 5% em água. O seu médico irá inverter gentilmente a bolsa para misturar a solução. Lemtrada não contém conservantes antimicrobianos e, portanto, o seu médico deve tomar cuidado para garantir a esterilidade da solução preparada. Cada frasco-ampola é para uso único. O produto diluído deve ser usado imediatamente após a diluição.
O seu médico irá solicitar exames de sangue antes de iniciar Lemtrada, continuando por quatro anos depois da última infusão de Lemtrada. É importante que você faça este exame de acordo com o esquema recomendado, para que o profissional de saúde possa observar os sinais e sintomas de efeitos colaterais autoimunes, de forma que o tratamento possa ser feito rapidamente, se necessário.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Lemtrada?
Se você não puder comparecer ao hospital ou a clínica médica para receber a dose de Lemtrada nas datas programadas, converse com seu médico.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Antes do tratamento, o seu médico deve informar de forma educativa sobre os riscos e os benefícios, bem como a necessidade de se comprometer com 48 meses de acompanhamento depois da última infusão de Lemtrada. Você deve observar os sintomas que podem ocorrer (veja seção abaixo) e imediatamente procure ajuda médica se tiver qualquer preocupação.
É importante que você siga a solicitação do seu profissional de saúde para realizar exames regulares de sangue e urina de forma que, se ocorrerem efeitos colaterais associados ao Lemtrada, eles possam ser reconhecidos cedo e tratados prontamente. É muito importante que você continue a realizar estes exames durante quatro anos após sua última infusão de Lemtrada, mesmo se você estiver se sentindo bem (sem sintomas ou efeitos colaterais), e se seus sintomas da esclerose múltipla estiverem sob controle. Uma vez que estes efeitos colaterais podem ocorrer muitos anos após seu tratamento com Lemtrada e podem (em casos raros) oferecer risco de vida, é muito importante que você continue a seguir a solicitação do profissional de saúde para realizar exames regulares de sangue e urina e observar o aparecimento de sintomas.
Autoimunidade
O sistema imunológico do seu corpo produz substâncias denominadas de anticorpos, que ajudam a combater as infecções. Os efeitos colaterais autoimunes são doenças que ocorrem quando o seu corpo produz anticorpos contra ele mesmo. Lemtrada pode fazer o seu corpo desenvolver anticorpos que têm como alvo certos órgãos, tais como a tireóide. Estes anticorpos podem levar ao desenvolvimento de efeitos colaterais como púrpura trombocitopênica idiopática (PTI ou nível baixo de plaquetas), distúrbios da tireoide ou, em casos raros, doenças nos rins. Ninguém pode prever quem irá desenvolver um efeito colateral autoimune. A realização de exames regulares de laboratório e estar ciente dos sinais e sintomas podem ajudar na detecção e no diagnóstico precoce, que podem proporcionar a melhor chance de melhora.
Púrpura trombocitopênica idiopática (PTI ou nível baixo de plaquetas)
Lemtrada pode causar uma doença conhecida como PTI, que resulta da diminuição do número de plaquetas no sangue. As plaquetas são necessárias para a coagulação normal do sangue. A PTI pode provocar sangramento sério que, se não tratado, pode levar a complicações graves da saúde e, possivelmente, à morte. Se detectada cedo, geralmente a PTI é tratável. O seu médico irá solicitar um exame de sangue antes de iniciar Lemtrada e uma vez por mês depois do ciclo inicial de tratamento, e continuar por mais quatro anos depois da última infusão de Lemtrada. Este exame de sangue ajudará o seu médico a observar as alterações na sua contagem de plaquetas a fim de identificar este efeito colateral precocemente. Importantemente, a PTI também pode ser detectada por certos sinais e sintomas que você precisa estar ciente, tais como: ocorrência de hematomas com facilidade, sangramento de um corte que é difícil de ser estancado, sangramento menstrual mais intenso do que o normal, sangramentos das gengivas ou do nariz de ocorrência nova ou que demoram mais que o normal para parar, manchas na pele pequenas e dispersas, vermelhas, cor de rosa ou violetas.
Entre em contato imediatamente com o seu médico se você apresentar algum destes sinais ou sintomas. Se não for possível encontrá-lo, procure atendimento médico imediatamente.
Distúrbios da tireoide
A tireoide é uma glândula encontrada na parte inferior do pescoço. Esta glândula produz hormônios que são importantes para o seu organismo todo. Lemtrada pode provocar o aparecimento de distúrbios da tireoide, incluindo uma glândula tireoide hiperativa ou hipoativa. Em geral, os distúrbios da tireoide são tratáveis, embora eles possam necessitar de tratamento para o resto da vida. O seu médico irá solicitar um exame de sangue antes de iniciar Lemtrada e a cada três meses depois do ciclo inicial do seu tratamento, continuando durante quatro anos depois da última infusão de Lemtrada.
Este exame de sangue irá auxiliar o profissional de saúde a detectar precocemente a doença da tireoide.
Entre em contato com o seu médico se você tiver:
Sintomas de uma tireóide hiperativa tais como transpiração excessiva, perda de peso sem explicação, inchaço do olho, nervosismo ou batimento rápido do coração ou sintomas de uma tireoide hipoativa tais como, ganho de peso sem explicação, sentir frio, piora do cansaço ou ocorrência de constipação nova.
Converse com seu médico se você estiver planejando ficar grávida ou se você engravidar depois de receber Lemtrada, pois a doença da tireoide não tratada pode prejudicar você ou o seu bebê em desenvolvimento.
Gravidez e Amamentação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Se você estiver grávida ou amamentando, suspeitar que está grávida ou estiver planejando engravidar, converse com o seu médico antes de tomar este medicamento. Não se sabe se Lemtrada pode prejudicar o bebê antes do nascimento. Você deve usar métodos de controle de natalidade eficazes durante o tratamento com Lemtrada e durante quatro meses depois de cada ciclo de tratamento com Lemtrada (exceto se o seu médico informá-la que isto não é necessário, pois você não pode ter filhos). Isto serve para garantir que não há Lemtrada remanescente no seu organismo, antes de você conceber uma criança.
Se você engravidar depois de receber o tratamento com Lemtrada e apresentar problemas de tireoide durante a gravidez, é necessário um cuidado extra. Os problemas de tireóide podem ser prejudiciais para o bebê.
Não se sabe se Lemtrada passa para o seu leite, mas isto acontece com muitos medicamentos. Você não deve amamentar durante cada ciclo de tratamento com Lemtrada e por quatro meses depois de cada ciclo de tratamento.
Uso em crianças
A segurança e a eficácia de Lemtrada em crianças com esclerose múltipla e menores de 18 anos de idade, não foram estabelecidas.
Uso em idosos
Os estudos clínicos de Lemtrada, não incluíram número suficiente de pacientes com 65 anos de idade ou mais, para determinar se eles respondem diferentemente dos pacientes mais jovens.
Comprometimento renal e hepático
Lemtrada não foi estudado em pacientes com comprometimento dos rins ou do fígado.
Abuso ou dependência da droga
Não há relatos de abuso ou dependência de Lemtrada pelos pacientes.
Interações medicamento-medicamento
Não foram conduzidos estudos formais de interação de Lemtrada com outros medicamentos, usando a dose recomendada em pacientes com esclerose múltipla. Portanto, Lemtrada não deve ser misturado com outros medicamentos na mesma veia.
Interação medicamento-alimento
Lemtrada é administrado por via intravenoso. Portanto, interações com alimentos e bebidas são improváveis.
Interação com exames de laboratório
Não se sabe se o alentuzumabe interfere em algum exame de laboratório clínico de rotina. Não há informações para sugerir que Lemtrada (alentuzumabe) possa causar doping. Em caso de dúvidas, consulte o seu médico.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Lemtrada pode causar efeitos colaterais sérios, incluindo efeitos colaterais autoimunes e infecções sérias:
- Dor de cabeça.
- Vômito.
- Erupção cutânea.
- Placas pruriginosas salientes na pele.
- Febre.
- Coceira.
- Náusea.
- Dificuldade para dormir.
- Tontura.
Outros efeitos colaterais comuns experimentados depois de um ciclo de tratamento incluem:
- Dor nas costas.
- Infecção do trato urinário.
- Aumento dos resfriados (ou na ocorrência de resfriado comum).
- Infecção do trato respiratório superior.
- Dor de garganta ou dor na boca.
- Infecção nos seios nasais.
- Dor em articulação.
- Hematomas na pele.
- Sensação de formigamento.
Informe seu médico se você apresentar algum efeito colateral que incomode ou que não vá embora.
Estes não são todos os possíveis efeitos colaterais de Lemtrada. Para mais informações, converse com o médico ou o farmacêutico.
Reação muito comum (ocorre em mais de 10% dos pacientes que usam este medicamento):
Náusea,diarreia, vômito, febre, cansaço; nasofaringite, infecção do trato urinário, infecção do trato respiratório, sinusite; contusão, dor nas costas, dor nas extremidades, artralgia (dor nas articulações); cefaleia, recidiva de esclerose múltipla, parestesia (sensação de queimação, cócegas ou espetadelas), tontura; insônia, dor na garganta; erupção cutânea, urticária e prurido.
Reação comum (ocorre entre 1% e 10% dos pacientes que usam este medicamento):
Linfopenia; taquicardia (batimento rápido do coração); dispepsia, dor abdominal; calafrios, desconforto no peito, dor, sintomas semelhantes aos da gripe, inchaço periférico; herpes oral, gripe, bronquite; linfócitos CD4 e CD8 diminuídos; fraqueza muscular, mialgia (dor muscular), espasmos musculares; disgeusia (alteração no paladar), hipoestesia (diminuição da sensação); depressão, ansiedade; tosse, dispnéia (encurtamento da respiração); erupção generalizada, eritema e ruborização.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
Composição
Cada mL contém:
10 mg de alentuzumabe.
Excipientes: edetato dissódico di-hidratado, cloreto de potássio, fosfato de potássio monobásico, cloreto de sódio, fosfato de sódio dibásico, polissorbato 80 e água para injetáveis.
Superdosagem
Não há antídoto conhecido para a superdose de alentuzumabe. O tratamento consiste de descontinuação do medicamento e administração de tratamento de suporte.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível.
Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Não foram conduzidos estudos formais de interação medicamentosa com Alentuzumabe (substância ativa) usando a dose recomendada em pacientes com EM. Em um estudo clínico controlado em EM, foi solicitado que os pacientes tratados recentemente com betainterferona e acetato de glatirâmer descontinuassem o tratamento 28 dias antes de iniciar o tratamento com Alentuzumabe (substância ativa).
Incompatibilidades farmacêuticas
Na ausência de estudos de compatibilidade, o alentuzumabe não deve ser misturado com outros medicamentos. Não adicionar ou administrar simultaneamente outros medicamentos por infusão, através da mesma via intravenosa.
Este medicamento não deve ser diluído com outros solventes além daqueles mencionados na seção “Como usar”.
Não há incompatibilidades conhecidas entre o alentuzumabe e bolsas de infusão de PVC (cloreto de polivinil), equipos de PVC ou de PVC revestidos com polietileno ou filtros de baixa afinidade por proteína.
Interação com exames de laboratório
Não se sabe se o alentuzumabe interfere em algum exame de laboratório clínico de rotina.
Interação Alimentícia
Alentuzumabe (substância ativa) é administrado por via parenteral. Portanto, interações com alimentos e bebidas são improváveis.
Ação da Substância
Resultados de eficácia
A segurança e a eficácia de Alentuzumabe (substância ativa) foram avaliadas em três estudos randomizados, avaliador-cego, com comparador ativo, em pacientes com EM.
Os Estudos 1 e 2 (CAMMS32400507 e CAMMS323) recrutaram pacientes com EM que haviam experimentado pelo menos dois episódios durante os dois anos anteriores. Exames neurológicos foram realizados a cada doze semanas e em tempos de suspeita de recidiva. Avaliações por ressonância magnética foram realizadas anualmente. Os pacientes foram acompanhados por dois anos. Em ambos estudos, os pacientes foram randomizados para receber Alentuzumabe (substância ativa) 12 mg/dia por infusão intravenosa (IV) administrada uma vez ao dia durante cinco dias no Mês 0 e por três dias no Mês 12 (grupo 12 mg) ou 44 mcg de IFNB-1a por injeção subcutânea (SC) administrada três vezes por semana.
O estudo 1 também incluiu um braço de dose exploratória para Alentuzumabe (substância ativa) 24 mg/dia, administrada uma vez ao dia durante cinco dias no Mês 0 e por três dias no Mês 12 (grupo 24 mg/dia). As medidas do desfecho primário para os Estudos 1 e 2 eram a taxa de recidiva anualizada (TRA) durante dois anos e o tempo para o início do “acúmulo sustentado de incapacidade” (ASI), definido como um aumento de pelo menos um ponto na escala expandida do estado de incapacidade (EDSS), a partir de uma pontuação de base ? 1,0 (aumento de 1,5 pontos para pacientes com a pontuação basal de EDSS igual a 0) que foi mantido por seis meses.
O Estudo 1 (CAMMS32400507) incluiu pacientes com esclerose múltipla recorrente remitente (EMRR) com EDSS de 0-5, com n = 426 no grupo Alentuzumabe (substância ativa) 12 mg e n = 202 no grupo IFNB-1a. A média de idade era 35 anos, a duração média da doença era de 4,5 anos e a pontuação média da EDSS era 2,7 no momento basal. Antes do recrutamento, os pacientes experimentaram pelo menos uma recidiva durante o tratamento com betainterferona ou acetato de glatirâmer, depois de terem sido tratados com estas drogas por pelo menos seis meses. No momento basal, a duração média de exposição às terapias anteriores para EM (? 1 droga usada) era de 35 meses no grupo Alentuzumabe (substância ativa) 12 mg; 29% haviam recebido ? 2 terapias anteriores para EM.
A TRA foi reduzida significantemente em 49% nos pacientes no grupo Alentuzumabe (substância ativa) 12 mg em comparação com o IFNB-1a SC durante dois anos. Além disso, o tratamento com Alentuzumabe (substância ativa) reduziu significantemente em 42% o risco de ASI em seis meses versus IFNB-1a SC durante dois anos. Os desfechos secundários principais incluíram a variação no escore de base da EDSS e dos parâmetros da ressonância magnética.
A pontuação média da EDSS foi significantemente reduzida em dois anos em pacientes tratados com Alentuzumabe (substância ativa), indicando uma melhora na pontuação da incapacidade, enquanto que a pontuação média da EDSS para os pacientes tratados com IFNB-1a aumentou significantemente em relação ao basal. Comparado com os pacientes tratados com IFNB-1a, os pacientes tratados com Alentuzumabe (substância ativa) tinham 2,6 vezes mais probabilidade de atingir uma redução sustentada na incapacidade. Os efeitos do tratamento nos resultados clínicos foram sustentados por efeitos significantes nas medidas da inflamação e progressão da doença por ressonância magnética, incluindo o volume do cérebro. Os resultados são mostrados na Tabela 1 e na Figura 1.
Tabela 1: Principais Resultados Clínicos e da Ressonância Magnética do Estudo 1:
| Resultado | ||
| Alentuzumabe (substância ativa) (N=426) | IFNB-1a SC (N=202) | |
| Resultados clínicos | ||
| Taxa de recidiva (desfecho co-primário) | ||
| TRA (95% IC) | 0,26 (0,21; 0,33) | 0,52 (0,41; 0,66) |
| Razão de taxas (95% IC) | 0,51 (0,39; 0,65) | |
| Valor-p | <0,0001 | |
| Incapacidade (ASI ?6 meses; desfecho co-primário) | ||
| Estimativa de pacientes com ASI em 6 meses (95% IC) | 12,71 (9,89; 16,27) | 21,13 (15,95; 27,68) |
| Razão de risco (95% IC) | 0,58 (0,38; 0,87) | |
| Valor-p | 0,0084 | |
| Proporção de pacientes sem recidiva no Ano 2 (%) | ||
| Estimativa (95% IC) | 65,38 (60,65; 69,70) | 46,70 (39,53; 53,54) |
| Valor-p | <0,0001 | |
| Variação na EDSS de base no Ano 2 | ||
| Estimativa (95% IC) | -0,17 (-0,29; -0,05) | 0,24 (0,07, 0,41) |
| Valor-p | <0,0001 | |
| Redução sustentada na incapacidade (RSI) | ||
| Estimativa de pacientes com RSI aos 6 meses (95% IC) | 28,82 (24,18; 34,13) | 12,93 (8,34; 19,77) |
| Razão de risco (95% IC) | 2,57 (1,57; 4,20) | |
| Valor-p | 0,0002 | |
| Resultados da Ressonância Magnética | ||
| Variação no volume da lesão em T2-RM do basal para o Ano 2 (%) | -1,27 | -1,23 |
| Valor-p | 0,1371 | |
| Pacientes com lesões novas ou aumentadas em T2 durante o Ano 2 (%) | 46,2 | 67,9 |
| Valor-p | <0,0001 | |
| Pacientes com lesões contrastadas com gadolinio durante o Ano 2 (%) | 18,5 | 34,2 |
| Valor-p | <0,0001 | |
| Pacientes com lesões novas pouco intensas em T1 durante o Ano 2 (%) | 19,9 | 38,0 |
| Valor-p | <0,0001 | |
| Alteração na Fração Parenquimatosa Cerebral basal para o Ano 2 (%) | -0,615 | |
| Valor-p | 0,0121 | -0,810 |
Variação média é apresentada para a EDSS. Variação mediana é apresentada para o volume da lesão em T2-RM e para a Fração Parenquimatosa Cerebral.
Figura 1: Tempo para o Acúmulo Sustentado de Incapacidade aos 6 Meses no Estudo 1:
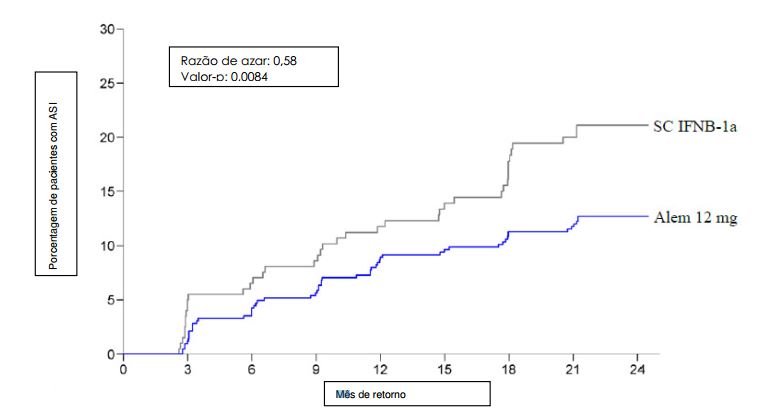
O Estudo 2 (CAMMS323) incluiu pacientes com EMRR, EDSS de 0-3,0, com n=376 no grupo Alentuzumabe (substância ativa) 12 mg e n=187 no grupo IFNB-1a. A média de idade era de 33 anos, a duração média da doença era de dois anos e a pontuação média da EDSS era de 2,0 no basal. Os pacientes não haviam recebido terapia anterior para EM na entrada do estudo.
A TRA foi significantemente reduzida em 55% nos pacientes tratados com Alentuzumabe (substância ativa) comparado com o IFNB-1a SC, em dois anos. Não houve diferença estatisticamente significante entre os grupos de tratamento no “acúmulo sustentado de incapacidade” em 6 meses; 8% dos pacientes tratados com Alentuzumabe (substância ativa) tinham aumento sustentado na pontuação da EDSS comparados com 11% dos pacientes com IFNB-1a.
Os efeitos do tratamento nos resultados clínicos foram suportados por efeitos significantes nas medidas da inflamação e progressão da doença por ressonância magnética, incluindo volume do cérebro. Os resultados são mostrados na Tabela 2.
Tabela 2: Principais Resultados Clínicos e da Ressonância Magnética do Estudo 2:
| Resultados | ||
| Alentuzumabe (substância ativa) (N=376) | IFNB-1a SC (N=187) | |
| Resultados clínicos | ||
| Taxa de recidiva (desfecho co-primário) | ||
| TRA (95% CI) | 0,18 (0,13; 0,23) | 0,39 (0,29; 0,53) |
| Razão de taxas (95% IC) | 0,45 (0,32; 0,63) | |
| Valor-p | <0,0001 | |
| Incapacidade (ASI ?6 meses; desfecho co-primário) | ||
| Estimativa de pacientes com ASI em 6 meses (95% IC) | 8,00 (5,66; 11,24) | 11,12 (7,32; 16,71) |
| Razão de risco (95% IC) | 0,70 (0,40; 1,23) | |
| Valor-p | 0,2173 | |
| Proporção de pacientes sem recidiva no Ano 2 (%) | ||
| Estimativa (95% IC) | 77,59 (72,87; 81,60) | 58,69 (51,12; 65,50) |
| Valor-p | <0,0001 | |
| Variação na EDSS do basal para o Ano 2 | ||
| Estimativa (95% IC) | -0,14 (-0,25; -0,02) | -0,14 (-0,29; 0,01) |
| Valor-p | 0,4188 | |
| Resultados da Ressonância Magnética | ||
| Variação no volume da lesão em T2-RM do basal para o Ano 2 (%) | -9,3 (-19,6; -0,2) | -6.5 (-20,7; 2,5) |
| Valor-p | 0,3080 | |
| Pacientes com lesões novas ou aumentadas em T2 durante o Ano 2 (%) | 48,5 | 57,6 |
| Valor-p | 0,0352 | |
|
Pacientes com lesões contrastadas com gadolinio durante o Ano 2 (%) | 15,4 | 27,0 |
| Valor-p | 0,0008 | |
| Pacientes com lesões novas pouco intensas em T1 durante o Ano 2 (%) | 24,0 | 31,4 |
| Valor-p | 0,0545 | |
| Variação na Fração Parenquimatosa Cerebral do basal para o Ano 2 (%) | -0,867 | -1,488 |
| Valor-p | <0,0001 | |
Variação média é apresentada para a EDSS. Variação mediana é apresentada para o volume da lesão em T2-RM e para a Fração Parenquimatosa Cerebral.
O Estudo 3 (CAMMS223) avaliou a segurança e a eficácia de Alentuzumabe (substância ativa) em pacientes com EMRR durante o curso de cinco anos. Os pacientes tinham um EDSS de 0-3,0, pelo menos dois episódios clínicos de EM nos dois anos anteriores e ?1 lesão contrastada por gadolínio na entrada no estudo. Os pacientes foram tratados com Alentuzumabe (substância ativa) 12 mg/dia (n=108) ou 24 mg/dia (n=108), administrado uma vez por dia durante cinco dias no Mês 0 e durante três dias no Mês 12 ou 44 mcg de IFNB-1a SC, administrado três vezes por semana durante três anos. Quarenta e seis pacientes receberam um terceiro ciclo planejado de tratamento com 12 mg/dia ou 24 mg/dia de Alentuzumabe (substância ativa) durante três dias no Mês 24.
Em três anos, Alentuzumabe (substância ativa) 12 mg reduziu o risco de ASI em seis meses em 76% (razão de risco 0,24 [95% IC: 0,110; 0,545], p>0,0006) e reduziu a TRA em 67% (razão de risco 0,33 [95% IC: 0,196, 0,552], p<0,0001) comparado com o IFNB-1a SC. Em cinco anos, Alentuzumabe (substância ativa) 12mg reduziu o risco de ASI em 69% (razão de risco 0,31 [95% IC: 0,161, 0,598], p=0,0005) e reduziu a TRA em 66% (razão de taxas 0,34 [95% IC: 0,202; 0569], p<0,0001) comparado com o IFNB-1a SC.
Características Físicas
Mecanismo de ação
Alentuzumabe (substância ativa) liga-se ao CD52, um antígeno da superfície celular presente em níveis altos em linfócitos T e B e em níveis menores em “células matadoras” (killer cells) naturais, monócitos e macrófagos. Há pouco ou nenhum CD52 detectado em neutrófilos, células plasmáticas ou células-tronco da medula óssea. Alentuzumabe (substância ativa) atua através da citólise celular dependente de anticorpo e lise mediada por complemento, depois da ligação à superfície celular de linfócitos T e B.
O mecanismo pelo qual Alentuzumabe (substância ativa) exerce seu efeito terapêutico na esclerose múltipla é desconhecido, mas pode envolver a imunomodulação através da depleção e repopulação de linfócitos. As pesquisas sugerem que os efeitos imunomoduladores potenciais na EM podem incluir alterações no número, proporções e propriedades de alguns subgrupos de linfócitos pós-tratamento.
Farmacodinâmica
Alentuzumabe (substância ativa) causa depleção de linfócitos T e B circulantes depois de cada ciclo de tratamento, com os menores níveis observados um mês após um ciclo de tratamento. A repopulação de linfócitos ocorre com o tempo, com recuperação completa das células B, geralmente dentro de seis meses.
As contagens de linfócitos T aumentam mais lentamente em direção ao normal, mas geralmente não retornam ao valor de base, até doze meses pós-tratamento. Aproximadamente 40% dos pacientes tinham contagens totais de linfócitos atingindo o limite inferior da normalidade em seis meses depois de cada ciclo de tratamento, e aproximadamente 80% dos pacientes apresentavam contagem total de linfócitos atingindo o limite inferior da normalidade até doze meses depois de cada ciclo de tratamento. Neutrófilos, monócitos, eosinófilos, basófilos e células matadoras naturais são afetados apenas transitoriamente por Alentuzumabe (substância ativa).
Farmacocinética
A farmacocinética de Alentuzumabe (substância ativa) foi avaliada em um total de duzentos e dezesseis pacientes com EMRR, que receberam infusões IV de 12 mg/dia ou 24 mg/dia por cinco dias consecutivos, seguido por três dias consecutivos, doze meses após o ciclo inicial de tratamento. As concentrações séricas aumentaram com cada dose consecutiva dentro de um ciclo de tratamento, com as maiores concentrações ocorrendo depois da última infusão do ciclo. A administração de 12 mg/dia resultou em uma Cmáx de 3014 ng/mL no Dia 5 do ciclo inicial de tratamento e 2276 ng/mL no Dia 3 do segundo ciclo de tratamento. A meia-vida alfa foi próxima de dois dias e comparável entre os ciclos, levando a concentrações séricas baixas ou não detectáveis dentro de aproximadamente trinta dias depois de cada ciclo de tratamento.
A farmacocinética da população de Alentuzumabe (substância ativa) foi melhor descrita por um modelo linear de dois compartimentos. A depuração sistêmica diminuiu com a contagem de linfócitos devido à perda do antígeno CD52 na periferia; entretanto, a diminuição do Ciclo 1 para o Ciclo 2 foi menos de 20%. O volume de distribuição central foi proporcional ao peso corporal e estava próximo do volume do líquido extracelular (14,1 L), sugerindo que Alentuzumabe (substância ativa) está amplamente confinado ao sangue e ao espaço intersticial. Não foi observado efeito da idade, raça ou gênero na farmacocinética de Alentuzumabe (substância ativa).
Cuidados de Armazenamento
Lemtrada deve ser conservado sob refrigeração (temperatura de 2ºC a 8ºC). Não congelar ou agitar. Proteger da luz.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após a diluição, utilizar o produto diluído imediatamente. Proteger da luz. Os frascos parcialmente usados, não usados ou danificados devem ser descartados de acordo com as políticas institucionais.
Características físicas
Lemtrada é uma solução estéril, concentrada (pH 7,0-7,4) límpida, incolor a levemente amarela para infusão.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS: 1.2543.0025
Farm. Resp.:
Bruna Belga Cathala - CRF-SP n° 42.670
Fabricado para:
Genzyme Corporation, Cambridge, USA
Por:
Boehringer Ingelheim Pharma GmbH & Co. KG
Biberach, Baden-Württemberg, Alemanha
Rotulado e embalado por: Genzyme Limited, Haverhill, Reino Unido
Ou
Fabricado para:
Genzyme Corporation, Cambridge, USA
Por: Boehringer Ingelheim Pharma GmbH & Co. KG
Biberach, Baden-Württemberg, Alemanha
Rotulado e embalado por:
Genzyme Ireland Limited, Waterford, Irlanda
Importado por:
Genzyme do Brasil Ltda.
Rua Padre Chico, 224 São Paulo – SP – CEP: 05008-010
CNPJ: 68.132.950/0001-03
Indústria Brasileira
SAC: 0800 77 123 73
www.genzyme.com.br
Venda sob prescrição médica.
informações complementares
| Fabricante |
| SANOFI |
| Princípio ativo |
| Alentuzumabe |
| Categoria do medicamento |
| Medicamentos Especiais |