Comparamos o preço de Xeloda - 500Mg C 120 Comprimidos Uso Restrito A Hospitais, veja o menor preço

R$ 2.200,00
RReferência
15
ofertasMelhores preços a partir de R$ 2.200,00 até R$ 3.471,00
Menor preço
vendido por Oncolog Medicamentos Especiais
economize
36.62%
R$ 2.200,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Aliria Medicamentos Especiais
economize
31.98%
R$ 2.361,00
vendido por Drogaria Dinâmica
economize
25.99%
R$ 2.568,72
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Facilita Medicamentos
economize
25.84%
R$ 2.573,97
vendido por OncoExpresso Medicamentos
economize
25.67%
R$ 2.580,00
vendido por Sampharma Medicamentos Especiais
economize
25.09%
R$ 2.600,00
vendido por Maranata Medicamentos
economize
7.17%
R$ 3.222,00
vendido por Pague Menos
economize
5.31%
R$ 3.286,65
vendido por Farma Visa
economize
4.96%
R$ 3.298,70
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Life Medicamentos
economize
4.96%
R$ 3.298,70
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Imune Farma Medicamentos Especiais
economize
4.96%
R$ 3.298,80
vendido por Farma Ame
economize
4.96%
R$ 3.298,85
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Fast Medicamentos
economize
4.09%
R$ 3.329,14
vendido por Farma Silva
economize
2.07%
R$ 3.399,00
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Saúde Farma Medicamentos
R$ 3.471,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
Para que serve
Xeloda é indicado para o tratamento de câncer de mama, câncer de cólon e reto (que são partes do intestino grosso) e câncer gástrico nas seguintes condições:
Câncer de mama:
- Xeloda em combinação com docetaxel é indicado para o tratamento de pacientes com câncer de mama com metástases (focos de células cancerosas distantes do foco primário), após falha da quimioterapia com antraciclina.
- Xeloda como tratamento único é indicado para o tratamento de pacientes com câncer de mama com metástases que não tenham apresentado resposta satisfatória a regimes de quimioterapia com paclitaxel e antraciclina ou para pacientes com resistência a paclitaxel e que não possam receber antraciclina, como pacientes que receberam doses cumulativas de 400 mg/m2 de doxorrubicina ou equivalente. Define-se resistência como progressão da doença na vigência do tratamento, com ou sem resposta inicial, ou recorrência em até 6 meses do término do tratamento adjuvante com antraciclina ou regimes com antraciclina.
Câncer colorretal:
- Xeloda é indicado para o tratamento adjuvante de pacientes com câncer colorretal.
- Xeloda é indicado como tratamento de primeira linha para pacientes com câncer de colorretal com metástases.
- Xeloda combinado com oxaliplatina ou combinado com oxilaplatina e bevacizumabe é indicado para tratamento de primeira linha de câncer colorretal metastático.
- Xeloda também pode ser combinado com oxaliplatina para o tratamento de segunda linha do câncer colorretal metastático em pacientes previamente tratados com irinotecano em combinação com um regime de fluoropirimidina como terapia de primeira linha.
Câncer gástrico:
-
Xeloda é indicado como tratamento de primeira linha para pacientes com câncer gástrico em estágio avançado, desde que associado com compostos de platina, como a cisplatina ou oxaliplatina.
Como Xeloda funcina?
Xeloda contém a substância ativa capecitabina que interrompe o crescimento das células tumorais ou cancerígenas (agente citostástico).
Contraindicação
Você não deve tomar Xeloda caso apresente alergia conhecida a qualquer um de seus componentes ou medicamentos à base de fluoropirimidinas e fluoruracila.
Você não poderá tomar Xeloda se for portador de deficiência de uma enzima chamada diidropirimidina desidrogenase.
Xeloda não deve ser administrado em conjunto com medicamentos como sorivudina e seus análogos ou com brivudina (medicamentos utilizados para o tratamento de herpes e catapora).
Este medicamento é contraindicado para pessoas que apresentem insuficiência renal grave (depuração de creatinina inferior a 30 mL/min).
Se existirem contraindicações para qualquer um dos agentes em combinação, o agente não deve ser utilizado.
Converse com o seu médico caso tenha dúvidas a respeito das possíveis contraindicações de Xeloda.
Como usar
Tomar os comprimidos por via oral, pela manhã e à noite, até 30 minutos após as refeições.
Ingerir os comprimidos com água.
Posologia
Seu médico prescreverá a dose adequada, dependendo da natureza de sua doença, de seu peso corpóreo e de sua resposta individual a Xeloda. Seu médico o informará sobre a quantidade correta de comprimidos que você deverá tomar pela manhã e à noite. Não mude as doses por sua conta. Em alguns casos, pode ser necessário reduzir a dose e seu médico saberá identificar essa situação para orientá-lo adequadamente.
Monoterapia
Câncer de mama e colorretal
A dose recomendada para monoterapia de Xeloda é 1.250 mg/m2, duas vezes ao dia (pela manhã e à noite; equivalente a 2.500 mg/m2 de dose total diária) durante 14 dias, seguidos de sete dias de pausa.
Terapia combinada
Câncer de mama
Em combinação com docetaxel, a dose recomendada de Xeloda é de 1.250 mg/m2, duas vezes ao dia (pela manhã e à noite, equivalente a 2.500 mg/m2 de dose total diária), durante 14 dias, seguidos de sete dias de pausa, associada ao docetaxel, 75 mg/m2, por infusão intravenosa, durante uma hora, a cada três semanas. A pré-medicação, de acordo com a bula de docetaxel, deve ser iniciada antes da administração de docetaxel para os pacientes que estiverem recebendo o medicamento em combinação com Xeloda.
Câncer colorretal e gástrico
No tratamento combinado, a dose inicial recomendada de Xeloda é de 800 a 1.000 mg/m2, administrada duas vezes ao dia durante duas semanas, seguida de período de sete dias de descanso, ou 625 mg/m2, duas vezes ao dia, quando administrada continuamente.
A inclusão de agentes biológicos em um esquema de associação não tem efeito sobre a dose inicial de Xeloda.
Pré-medicação para manter controlada a hidratação e antiemese, como descrito na bula da cisplatina e oxaliplatina, deve ser iniciada antes da administração de cisplatina para os pacientes que forem submetidos ao tratamento de Xeloda em combinação com cisplatina ou oxaliplatina.
Poderá haver necessidade de ajustes da dose em casos de insuficiência renal, de toxicidade ou durante tratamento em associação com outros quimioterápicos. Informe seu médico sobre o aparecimento de reações desagradáveis.
Instruções especiais de doses
Pacientes com insuficiência hepática devida a metástases hepáticas:
Se a insuficiência hepática é leve a moderada, nenhum ajuste da dose inicial é necessário. Nesses casos os pacientes devem ser cuidadosamente monitorados. Não foram estudados pacientes com insuficiência hepática grave.
Pacientes com insuficiência renal:
Em pacientes com insuficiência renal moderada, recomenda-se uma dose inicial menor, conforme orientação médica. Em pacientes com insuficiência renal leve, não se recomendam ajustes da dose inicial. Esses pacientes devem ser cuidadosamente monitorados por seus médicos. A recomendação de ajuste de dose para pacientes com insuficiência renal moderada se aplica tanto à monoterapia quanto ao uso em combinação.
Crianças:
A segurança e a eficácia de Xeloda em crianças não foram estabelecidas.
Idosos:
Para a monoterapia de Xeloda não são necessários ajustes da dose inicial. No entanto, recomenda-se monitoramento cuidadoso dos pacientes idosos em relação às reações adversas graves (grau 3 ou 4).
Em combinação com docetaxel, foi observada incidência aumentada de eventos adversos (grau 3 ou 4) e de eventos adversos graves em pacientes com 60 anos de idade ou mais. Recomenda-se a redução da dose inicial de Xeloda para 75% (950 mg/m2 duas vezes ao dia) em pacientes com 60 anos de idade ou mais, conforme orientação médica.
Duração do tratamento
A duração do tratamento com Xeloda varia, dependendo da natureza de sua doença e de sua resposta individual ao tratamento. Seu médico o informará sobre quando você deve parar de tomar Xeloda.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que devo fazer quando eu me esquecer de usar Xeloda?
Caso você esqueça de usar o medicamento, não tome uma dose extra. Aguarde até a dose seguinte e tome a sua dose normal em seguida.
Não tente compensar a dose que você esqueceu tomando mais de uma dose ao mesmo tempo. As doses não tomadas de Xeloda, devido à toxicidade, não são substituídas.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Enquanto você estiver tomando Xeloda haverá necessidade de acompanhamento médico cuidadoso.
Embora a maioria dos efeitos colaterais seja reversível, pode ser necessário suspender a medicação ou reduzir a dose em alguns casos.
Prisão de ventre, boca seca e gases são eventos gastrintestinais comuns à terapia combinada de Xeloda com outras medicações, como a oxaliplatina.
Xeloda pode induzir diarreia, que pode ser grave. Se você apresentar diarreia grave, deverá ser acompanhado cuidadosamente e, se ficar desidratado, deve receber fluidos com reposição de eletrólitos. Tratamentos para a diarreia devem ser iniciados o quanto antes, quando indicado. A desidratação precisa ser evitada ou corrigida logo no início.
Os pacientes com perda de apetite, diminuição da força muscular acompanhada de fraqueza, náusea, vômito ou diarreia podem ficar desidratados rapidamente.
Desidratação pode causar insuficiência renal aguda, especialmente em pacientes que já apresentem comprometimento da função renal ou quando Xeloda é administrado junto com outros medicamentos tóxicos para os rins.
Casos de falência renal seguidos de morte foram reportados nessas situações. Se a desidratação for grave, o tratamento com Xeloda precisará ser interrompido, até que você se recupere totalmente.
Foi observada toxicidade ao coração com o uso de Xeloda, incluindo infarto do miocárdio, angina, arritmias, parada cardíaca, insuficiência cardíaca e alterações no eletrocardiograma. Esses eventos adversos podem ser mais comuns em pacientes que já apresentavam doença das artérias coronárias anteriormente.
Xeloda pode provocar reações de pele graves, como síndrome de Stevens-Johnson (inclui lesões cutâneas generalizadas, como bolhas, que podem atingir também as mucosas) e necrólise epidérmica tóxica (camada superficial da pele se solta em lâminas). Xeloda deve ser permanentemente descontinuado nesses casos.
Xeloda pode provocar a síndrome mão-pé, uma lesão de pele com gravidade variável (grau 1 a 3), em média 79 dias depois do início do tratamento, com uma variação de 11 a 360 dias. Síndrome mão-pé persistente ou grave (grau 2 ou maior) pode, eventualmente, levar à perda de impressões digitais, o que poderia impactar a identificação do paciente.
No grau 1, aparece formigamento nas mãos e nos pés, acompanhado de vermelhidão, mas o paciente consegue continuar com suas atividades. No grau 2, mãos e pés ficam muito doloridos e inchados, além de vermelhos, e o paciente já não consegue realizar suas atividades normalmente. No grau 3, aparecem feridas e bolhas, a pele se descola, e o desconforto é muito grande. Se a síndrome for de grau 2 ou 3, o tratamento com Xeloda precisa ser interrompido até a resolução ou melhora do quadro.
Há evidências de que dexpantenol funciona na prevenção da síndrome mão-pé.
Xeloda pode induzir a aumento das bilirrubinas (substâncias produzidas pelo fígado que, quando aumentadas, podem levar ao aparecimento de cor amarelada na pele e nos olhos).
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas.
Não foram realizados estudos sobre os efeitos de Xeloda sobre a capacidade de dirigir veículos ou operar máquinas.
Gravidez e amamentação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Não foram realizados estudos com mulheres grávidas em uso de Xeloda.
No entanto, com base no mecanismo de ação deste medicamento, que interrompe a multiplicação das células, presume-se que Xeloda possa causar dano para o feto se administrado a mulheres grávidas.
Antes de iniciar o tratamento, você deve informar ao seu médico caso pretenda engravidar.
Você não deve tomar Xeloda caso esteja grávida ou pense que poderia estar. Você não deve amamentar caso esteja tomando Xeloda.
Populações especiais
Pacientes idosos e pacientes com funções renal ou hepática comprometidas devem ser cuidadosamente monitorados, pois podem apresentar maior probabilidade de desenvolver quadros de toxicidade gastrintestinal, além de quadros de toxicidade mais grave.
Até o momento, não há informações de que Xeloda possa causar doping. Em caso de dúvidas, consulte o seu médico.
Principais interações medicamentosas
Anticoagulantes:
Avise seu médico se estiver tomando anticoagulantes como varfarina e femprocumona, pois o uso desses medicamentos em combinação com Xeloda pode alterar a coagulação.
Fenitoína:
Se você estiver recebendo fenitoína (medicamento usado para controlar convulsões) ao mesmo tempo que Xeloda, seu médico deve lhe monitorar regularmente as concentrações sanguíneas de fenitoína, que podem provocar efeitos colaterais.
Alimentos:
Em todos os estudos feitos com Xeloda, os pacientes foram instruídos a tomar Xeloda até 30 minutos após uma refeição. Portanto, recomenda-se que Xeloda seja administrado dessa forma.
Antiácidos:
Antiácidos contendo hidróxido de alumínio e hidróxido de magnésio podem causar um pequeno aumento nas concentrações plasmáticas de Xeloda.
Ácido folínico:
A toxicidade de Xeloda pode ser aumentada com o uso de ácido folínico.
Sorivudina e análogos:
Xeloda não deve ser administrado com sorivudina ou com seus análogos quimicamente semelhantes, como brivudina, pois existe o risco de aumentar a toxicidade de fluoropirimidinas e isso pode ser fatal. É necessário aguardar pelo menos 4 semanas entre o fim da terapia com soruvudinas ou medicamentos semelhantes e o início da terapia com Xeloda.
Alterações nos resultados de exames laboratoriais
Xeloda pode causar alterações nos exames laboratoriais, assim os pacientes devem realizar exames periodicamente durante o tratamento. O seu médico saberá como proceder adequadamente nesses casos.
Interrupção do tratamento
Seu médico pode solicitar que você interrompa o tratamento com Xeloda durante algum tempo ou que tome menor quantidade do medicamento, caso desenvolva qualquer reação adversa de difícil controle.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Além dos efeitos benéficos de Xeloda, é possível que ocorram efeitos indesejados durante o tratamento, mesmo quando usado conforme a prescrição médica.
Os efeitos indesejados comumente ocorrem no início do tratamento. Esses efeitos colaterais normalmente melhoram rapidamente em 2-3 dias. Se o tratamento com Xeloda for interrompido; o tratamento poderá ser reiniciado, de acordo com as instruções de seu médico.
Nos casos de diarreia com mais de quatro evacuações por dia e diarreia durante a noite, de vômitos mais de uma vez em 24 horas, ou se os sintomas nas mãos e pés se agravarem com presença de dor, inchaço ou bolhas ou ainda se a quantidade de alimentos que você ingere por dia está muito abaixo da normal e as feridas na boca se tornarem doloridas, pare de tomar Xeloda imediatamente e procure seu médico para obter orientação adicional.
Reações adversas de acordo com a indicação
Xeloda em monoterapia
| Reação adversa por sistema | Muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento) | Comum (ocorre entre 5 e 10% dos pacientes que utilizam este medicamento) |
| Distúrbios do metabolismo e nutrição | Perda de apetite | Desidratação, Diminuição do apetite |
| Distúrbios do sistema nervoso | Dormência ou sensações de formigamento, Alteração do paladar, Dor de cabeça Tontura (sem vertigem) | |
| Distúrbios oculares | Aumento do lacrimejamento, Conjuntivite | |
| Distúrbios gastrintestinais | Diarreia, Vômito, Náusea, Estomatite (feridas na boca),Dor abdominal (dor na barriga) | Prisão de ventre,Dor abdominal,Dificuldade de digestão |
| Distúrbios hepatobiliares | Excesso de bilirrubina no sangue | |
| Distúrbios da pele e tecido subcutâneo | Inchaço, vermelhidão, formigamento e adormecimento das palmas das mãos e plantas dos pés (síndrome mão-pé)* Dermatite | Erupções na pele, Perda de cabelo, Cor vermelha na pele,Pele seca |
| Distúrbios gerais e relacionados ao local de administração | Cansaço, Sono profundo | Febre, Fraqueza, Diminuição da força muscular acompanhada de fraqueza |
* Baseado na experiência pós-comercialização, síndrome mão-pé persistente ou grave pode eventualmente levar à perda de impressões digitais.
Reações adversas relatadas em menos de 5% dos pacientes tratados com Xeloda em monoterapia
Distúrbios gastrintestinais:
Boca seca, gases, reações adversas relacionadas à ulceração/inflamação de mucosas, como inflamação do esôfago, estômago, intestino delgado, intestino grosso e hemorragia (sangramento) gastrintestinal.
Distúrbios cardíacos:
Inchaço nas pernas, dor no peito de origem cardíaca, incluindo angina de peito, doença do músculo cardíaco, infarto/isquemia miocárdica, insuficiência cardíaca, morte súbita, aumento da frequência cardíaca, arritmias cardíacas e palpitações.
Distúrbios do sistema nervoso:
Insônia, confusão, comprometimento das funções cerebrais e sinais cerebelares, como falta de coordenação motora, dificuldade para articular as palavras, alteração no equilíbrio e alteração na coordenação.
Infecções e infestações:
Infecções locais, infecções generalizadas fatais (incluindo origem bacteriana, viral e fúngica) e sepse (infecção disseminada).
Distúrbios do sangue e do sistema linfático:
Anemia e redução de todas as células do sangue.
Distúrbios da pele e tecido subcutâneo:
Coceira, descolamento da pele localizado, escurecimento da pele, distúrbios das unhas, reações de sensibilidade à luz e sensibilidade à radioterapia.
Distúrbios gerais relacionados ao local de administração:
Dor nas pernas e braços e dor no peito (não cardíaca).
Olhos:
Irritação nos olhos.
Respiratórios:
Falta de ar e tosse.
Musculoesqueléticos:
Dor lombar, dor nos músculos e articulações.
Distúrbios psiquiátricos:
Depressão.
Insuficiência hepática e hepatite foram relatadas durante os estudos clínicos e após a comercialização, mas não foi estabelecida relação de causa com o tratamento com Xeloda.
Fissuras na pele (rachaduras) foram relatadas em menos que 2% dos pacientes em estudos clínicos de Xeloda.
Xeloda em terapia combinada
Reações adversas muito comuns e comuns com Xeloda em combinação com diferentes quimioterápicos
| Reação adversa por sistema | Muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento) | Comum (ocorre entre 5 e 10% dos pacientes que utilizam este medicamento) |
| Infecções e infestações | Diminuição das células brancas do sangue com ou sem febre, Diminuição das plaquetas, Anemia | Comum (ocorre entre 5 e 10% dos pacientes que utilizam este medicamento) |
| Distúrbios do sistema sanguíneo e linfático | Diminuição do apetite | Infecção Candidíase oral (sapinho) |
| Distúrbios do metabolismo e nutrição | Diminuição do cálcio no sangue, Diminuição de peso | |
| Distúrbios psiquiátricos | Insônia | |
| Distúrbios do sistema nervoso | Alteração dos nervos responsáveis pela sensibilidade de mãos e pés, Distúrbio no paladar, Sensibilidade alterada (dormência ou formigamentos), Dor de cabeça | Diminuição de sensibilidade |
| Distúrbios oculares | Aumento do lacrimejamento | |
| Distúrbios vasculares | Trombose / embolismo (entupimento de vasos sanguíneos por coágulos) Pressão alta, Inchaço nas pernas | |
| Respiratório | Dor na garganta | Sangramento pelo nariz, Rouquidão, Coriza, Falta de ar |
| Distúrbios gastrintestinais | Prisão de ventre, Dificuldade de digestão | Boca seca |
| Distúrbios da pele e tecido subcutâneo | Perda de cabelo, Alterações das unhas | Dor no maxilar, Dor nas costas |
| Distúrbios musculoesqueléticos e dos tecidos conectivos | Dores nas juntas, Dores musculares, Dores nos braços e pernas | |
| Desordens gerais e do local de administração | Febre, Diminuição da força muscular acompanhada de fraqueza Fraqueza, Intolerância à temperatura | Febre, Dor |
Reações de hipersensibilidade e isquemia/infarto do miocárdio foram comumente relatadas com o uso de Xeloda em combinação com outros quimioterápicos, mas em menos de 5% dos pacientes.
Reações adversas raras ou incomuns relatadas com Xeloda em combinação com outros quimioterápicos são compatíveis com as reações adversas descritas com o uso de Xeloda em monoterapia ou dos produtos combinados em monoterapia.
Pós-comercialização
Reações adversas a drogas (RADs) identificadas durante a exposição pós-comercialização
| Classe de sistemas e órgãos | Reações adversas a drogas | Frequência |
| Distúrbios renais e urinários | Insuficiência renal aguda secundária à desidratação | Rara (ocorre entre 0,01% e 0,1% dos pacientes que utilizam este medicamento) |
| Distúrbios no sistema nervoso | Leucoencefalopatia tóxica (danos ao sistema nervoso central, desencadeados por um agente químico) | Desconhecida |
| Distúrbios hepatobiliares | Insuficiência hepática, hepatite | Muito rara (ocorre em menos de 0,01% dos pacientes que utilizam este medicamento) |
| Distúrbios metabólicos e nutricionais | Hipertrigliceridemia (aumento da concentração de triglicerídeos) | Desconhecida |
| Distúrbios no tecido subcutâneo e pele | Lúpus eritematoso cutâneo (doença imunológica), reações de pele graves como Síndrome de Stevens-Johnson (doença com lesões cutâneas generalizadas, como bolhas, que podem atingir também as mucosas) e necrólise epidérmica tóxica (doença que acomete a camada superficial da pele e essa se solta em lâminas) | Muito rara (ocorre em menos de 0,01% dos pacientes que utilizam este medicamento) |
| Distúrbios nos olhos | Estenose do ducto lacrimal (estreitamento do canal lacrimal), distúrbios de córnea incluindo ceratite (inflamação da córnea) |
Atenção: Este produto é um medicamento que possui nova indicação terapêutica no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer reações adversas imprevisíveis ou desconhecidas. Nesse caso, informe seu médico.
Composição
Cada comprimido revestido de Xeloda 150 mg contém:
Capecitabina - 150 mg.
Cada comprimido revestido de Xeloda 500 mg contém:
Capecitabina - 500 mg.
Excipientes: Lactose anidra, croscarmelose sódica, hipromelose, celulose microcristalina, estearato de magnésio e água purificada. Componentes do revestimento: hipromelose, talco, dióxido de titânio, óxido de ferro amarelo, óxido férrico (óxido de ferro vermelho) e água purificada.
Superdosagem
As manifestações agudas de superdose (quantidade maior que a indicada) incluem náusea, vômitos, diarreia, inflamação das mucosas, irritação e sangramento gastrintestinal e diminuição na produção de células do sangue.
Procure imediatamente um médico em caso de superdose.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Anticoagulantes cumarínicos
Parâmetros de coagulação e/ou sangramento alterados foram relatados em pacientes que utilizavam concomitantemente capecitabina e anticoagulantes derivados da cumarina como a varfarina e femprocumona. Esses eventos ocorreram dentro de alguns dias e até alguns meses após o início da terapia com capecitabina e, em alguns casos, um mês após a interrupção da ingestão de capecitabina.
Em um estudo clínico de interação, após uma dose única de 20 mg de varfarina, capecitabina aumentou em 57% a AUC da S-varfarina, com um aumento de 91% do valor de INR. Esses resultados sugerem uma interação, provavelmente devida a uma inibição do sistema citocromo P450, isoenzima 2C9, por capecitabina.
Pacientes que tomam anticoagulantes derivados da cumarina concomitantemente com capecitabina devem ser monitorados regularmente em relação às alterações nos seus parâmetros de coagulação (TP ou INR), e a dose de anticoagulante deve ser ajustada apropriadamente.
Substratos do citocromo P450 2C9
Não foram realizados estudos formais de interação medicamentosa com capecitabina e outros fármacos metabolizados pelo citocromo P450 isoenzima 2C9. Devem ser adotadas precauções quando capecitabina for coadministrada com esses fármacos.
Fenitoína
O aumento na concentração plasmática de fenitoína foi relatado durante o uso concomitante com capecitabina. Não foram realizados estudos formais de interação medicamentosa com fenitoína, mas presume-se que o mecanismo de interação seja a inibição do CYP isoenzima 2C9 pela capecitabina.
Pacientes que recebem fenitoína concomitantemente com capecitabina devem ser regularmente monitorados quanto ao aumento das concentrações plasmáticas de fenitoína.
Antiácidos
O efeito dos antiácidos contendo hidróxido de alumínio e hidróxido de magnésio sobre a farmacocinética de capecitabina foi investigado em pacientes com câncer. Houve um pequeno aumento nas concentrações plasmáticas de capecitabina e do metabólito 5'-DFCR; não houve nenhum efeito nos três principais metabólitos (5'-DFUR, 5-FU e FBAL).
Leucovorin (ácido folínico)
Foi investigado o efeito de Leucovorin sobre a farmacocinética de capecitabina em pacientes com câncer. Leucovorin não tem nenhum efeito na farmacocinética da capecitabina e de seus metabólitos. Entretanto, apresenta um efeito sobre a farmacodinâmica de capecitabina, que pode ter sua toxicidade aumentada por Leucovorin.
Sorivudina e análogos
Foi descrita na literatura uma interação clinicamente significante entre sorivudina e 5-FU, resultante da inibição da diidropirimidina desidrogenase pela sorivudina. Essa interação acarreta aumento da toxicidade das fluoropirimidinas, que é potencialmente fatal. Portanto, capecitabina não deve ser administrada concomitantemente com sorivudina ou com seus análogos quimicamente relacionados, como a brivudina.
Deve haver um período de espera de, no mínimo, quatro semanas entre o fim da terapia com sorivudinas ou com seus análogos relacionados, como a brivudina, e o início da terapia com capecitabina.
Oxaliplatina
Não houve diferenças clinicamente significativas na exposição à capecitabina ou seus metabólitos, livres de platina ou platina total quando capecitabina e oxaliplatina foram administradas em combinação, com ou sem bevacizumabe.
Bevacizumabe
Não houve efeito clinicamente significativo de bevacizumabe nos parâmetros farmacocinéticos de capecitabina ou seus metabólitos.
Interação Alimentícia
Em todos os estudos clínicos, os pacientes foram instruídos a tomar capecitabina até 30 minutos após uma refeição. Considerando que os dados de segurança e de eficácia atuais são baseados na administração com alimentos, recomenda-se que capecitabina seja administrada com alimentos.
Ação da Substância
Resultados de eficácia
Câncer colorretal
Monoterapia em câncer colorretal adjuvante
Um estudo clínico controlado de fase III, multicêntrico, randomizado, em pacientes com câncer colorretal em estágio III (Dukes C) foi conduzido para estudar o uso de capecitabina como tratamento adjuvante de pacientes com câncer colorretal (Estudo X-ACT: M66001).
Nesse estudo, 1.987 pacientes foram randomizados para receberem tratamento com capecitabina (1.250 mg/m2, duas vezes ao dia, durante duas semanas, seguido por período de descanso de uma semana, e administrado em ciclos a cada três semanas, durante 24 semanas), ou 5-FU e Leucovorin (ácido folínico) (esquema de tratamento Mayo: 20 mg/m2 de Leucovorin intravenoso (i.v.), seguido por 425 mg/m2 i.v. em bolus de 5-FU, nos dias 1 a 5, a cada 28 dias durante 24 semanas).
Capecitabina foi pelo menos equivalente ao 5-FU/LV i.v. na sobrevida livre de doença (p = 0,0001, margem de não inferioridade de 1,2).
Em toda a população randomizada, as diferenças entre capecitabina e 5-FU/LV na sobrevida livre da doença e sobrevida global mostraram razão de risco de 0,88 (IC 95%, 0,77 – 1,01; p = 0,068) e 0,86 (0,74 – 1,01; p = 0,060), respectivamente. O tempo mediano de acompanhamento no momento da análise era de 6,9 anos.
Terapia combinada em câncer colorretal adjuvante
Capecitabina em combinação com oxaliplatina (Xelox) para o tratamento adjuvante de pacientes com câncer colorretal foi estudada em estudo clínico multicêntrico, randomizado, controlado de fase III, em pacientes com câncer colorretal estágio III (Dukes C) (estudo NO16968).
Nesse estudo, 944 pacientes foram randomizados para ciclos de três semanas, durante 24 semanas, com capecitabina (1.000 mg/m2, duas vezes ao dia, durante duas semanas, seguido por período de descanso de uma semana), em combinação com oxaliplatina (infusão intravenosa de 130 mg/m2 durante duas horas do dia 1, a cada três semanas) e 942 pacientes foram randomizados para bolus de 5-FU e Leucovorin.
Na análise primária para sobrevida livre de doença, na população com intenção de tratamento (ITT), Xelox mostrou-se significativamente superior a 5-FU/LV (razão de risco = 0,80, IC 95% = [0,69; 0,93], p = 0,0045). A taxa da sobrevida livre de doença de três anos foi de 71% para Xelox versus 67% para 5-FU/LV.
A análise para o objetivo secundário de sobrevida livre de recorrência apoia esses resultados com uma razão de risco de 0,78 (IC 95% = [0,67; 0,92], p = 0,0024) para Xelox versus 5-FU/LV. Xelox mostrou uma tendência a sobrevida global superior, com uma razão de risco de 0,87 (IC 95%= [0,72; 1,05], p = 0,1486) que se traduz em uma redução de 13% no risco de morte.
A taxa de sobrevida global de cinco anos foi de 78% para Xelox versus 74% para 5-FU/LV. Os resultados de eficácia baseiam-se em um tempo mediano de observação de 59 meses para a sobrevida global e 57 meses para a sobrevida livre de doença.
A taxa de retirada do estudo devido a eventos adversos foi maior no braço de terapia combinada de capecitabina com oxaliplatina (21%) quando comparada com ao braço 5-FU/LV (9%) na população ITT, segundo estudo NO16968.
Monoterapia em câncer colorretal metastático
Dois estudos clínicos com desenho idêntico, multicêntricos, randomizados, controlados, de fase III foram conduzidos para estudar o uso de capecitabina como tratamento de primeira linha no câncer colorretal metastático (SO14695; SO14796).
Nesses estudos, 603 pacientes foram randomizados para o tratamento com capecitabina (1.250 mg/m2, duas vezes ao dia, durante duas semanas, seguido de período de descanso de uma semana, em ciclos de três semanas) e 604 pacientes foram randomizados para o tratamento com 5- FU e Leucovorin (esquema de tratamento Mayo: 20 mg/m2 de Leucovorin i.v., seguido de 425 mg/m2 i.v. de 5-FU em bolus, nos dias 1 a 5, a cada 28 dias).
Os índices de resposta objetiva global em toda a população randomizada (avaliação do pesquisador) foi 25,7% (capecitabina) versus 16,7% (esquema de tratamento Mayo); p< 0,0002. O tempo mediano até a progressão foi de 140 dias (capecitabina) versus 144 dias (esquema de tratamento Mayo). A sobrevida mediana foi de 392 dias (capecitabina) versus 391 dias (esquema de tratamento Mayo).
Terapia combinada no tratamento de primeira linha de câncer colorretal
Um estudo clínico multicêntrico, randomizado, controlado, de fase 3 (NO16966) foi conduzido para o uso de capecitabina em combinação com oxaliplatina ou em combinação com oxaliplatina e bevacizumabe (BV) para o tratamento de primeira linha de câncer colorretal metastático.
O estudo foi composto por duas partes: uma parte inicial com 2 braços na qual os pacientes foram randomizados para dois grupos diferentes de tratamento, incluindo Xelox ou Folfox-4, e uma parte subsequente fatorial 2x2 com 4 grupos distintos de tratamento, incluindo Xelox + placebo (P), Folfox-4 + P, Xelox + BV e Folfox-4 + BV. Os regimes de tratamento estão resumidos na tabela a seguir:
Tabela 1. Regimes de tratamento no estudo NO16966
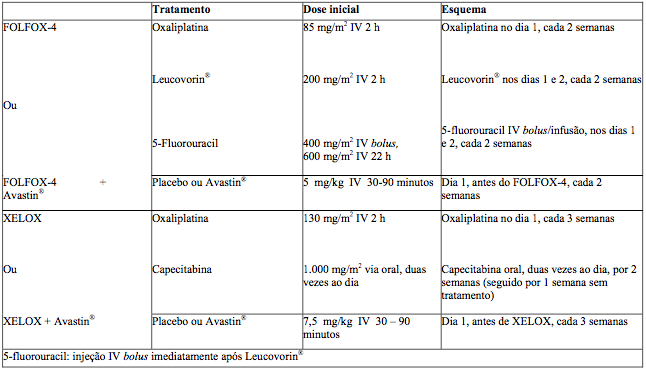
A não inferioridade dos braços contendo Xelox comparado aos braços contendo Folfox-4, na comparação global, foi demonstrada nos termos de sobrevida livre de progressão (SLP) na população de pacientes elegível e na população com intenção de tratamento (ITT) (veja tabela a seguir).
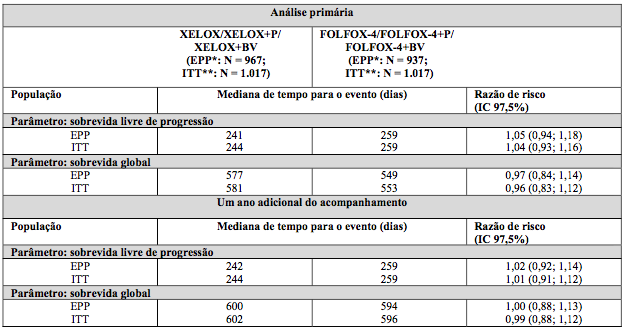
Os resultados indicam que Xelox é equivalente a Folfox-4 no que se refere à sobrevida global. Uma comparação de Xelox + bevacizumabe versus Folfox-4 + bevacizumabe foi uma análise exploratória pré-especificada. Na comparação desse subgrupo de tratamento, Xelox + bevacizumabe foi similar a Folfox-4 + bevacizumabe no que se refere a sobrevida livre de progressão [razão de risco 1,01 (IC 97,5% 0,84, 1,22)].
O acompanhamento mediano no tempo de análise primária na população com intenção de tratamento foi 1,5 anos. Os dados provenientes de análises após um ano adicional do acompanhamento também foram incluídos na tabela a seguir.
Tabela 2. Resultados de não inferioridade fundamentais para a análise primária e dos dados de acompanhamento de um ano (populações EPP e ITT, estudo NO16966)
 * EPP = população de pacientes elegíveis; ** ITT = população com intenção de tratamento.
* EPP = população de pacientes elegíveis; ** ITT = população com intenção de tratamento.
Terapia combinada no tratamento de segunda linha de câncer colorretal
O estudo clínico NO16967 fase 3, multicêntrico, randomizado e controlado estudou a utilização de capecitabina em combinação com oxaliplatina para o tratamento de segunda linha de câncer colorretal metastático.
Nesse estudo, 627 pacientes com carcinoma colorretal metastático, e previamente tratados com irinotecano em combinação com um regime de fluoropirimidina como terapia de primeira linha, foram randomizados para tratamento com Xelox ou Folfox-4.
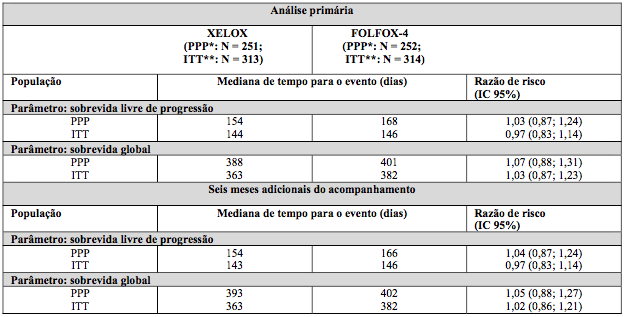
Para o esquema de dose de Xelox e Folfox- 4 (sem adição de placebo ou bevacizumabe), vide a Tabela 1, Xelox demonstrou ser não inferior a Folfox-4 em termos de sobrevida livre de progressão na população per-protocolo e na população com intenção de tratamento (vide a tabela a seguir).
Os resultados indicam que Xelox é equivalente a Folfox-4 em termos de SG. O acompanhamento mediano até o tempo da análise primária na população com intenção de tratamento foi 2,1 anos. Os dados provenientes de uma análise após seis meses adicionais do acompanhamento também estão incluídos na tabela a seguir.
Tabela 3. Resultados de eficácia / não inferioridade fundamentais para a análise primária e dos dados de acompanhamento de seis meses do Estudo NO16967 (populações PPP e ITT)
* PPP = população per-protocolo; ** ITT = população com intenção de tratamento.
Uma análise combinada dos dados de eficácia do tratamento de primeira-linha (estudo NO16966; parte inicial de 2 braços) e do tratamento de segunda-linha (estudo NO16967) proporciona suporte adicional aos resultados de não inferioridade de Xelox versus Folfox-4, conforme obtido nos estudos individuais: a sobrevida livre de progressão na população per-protocolo [razão de risco 1,00 (IC 95%: 0,88; 1,14)] com uma mediana de sobrevida livre de progressão de 193 dias (Xelox; 508 pacientes) versus 204 dias (Folfox-4; 500 pacientes).
Os resultados indicam que Xelox é equivalente a Folfox-4 em termos de SG [razão de risco 1,01 (IC 95%: 0,87; 1,17)] com uma mediana de SG de 468 dias (Xelox) versus 478 dias (Folfox-4).
Câncer gástrico
Terapia combinada
Um estudo clínico multicêntrico, randomizado, controlado, de fase III estudou a utilização de capecitabina para o tratamento de pacientes com câncer gástrico metastático ou avançado. Nesse estudo, 160 pacientes foram randomizados para tratamento com capecitabina (1.000 mg/m2, duas vezes ao dia, por duas semanas, seguido por período de descanso de uma semana) e cisplatina (80 mg/m2 em infusão de duas horas, a cada três semanas).
Um total de 156 pacientes foram randomizados para tratamento com 5-FU (800 mg/m2 por dia, infusão contínua dos dias 1 ao 5, durante três semanas) e cisplatina (80 mg/m2, em infusão no dia 1, a cada três semanas).
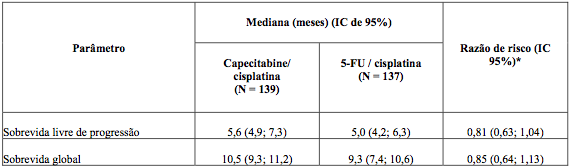
O objetivo primário do estudo foi alcançado, capecitabina foi ao menos equivalente ao 5-FU em combinação com cisplatina no que se refere à sobrevida livre de progressão (análise realizada na população per-protocolo).
O resultado da sobrevida global foi similar ao resultado da sobrevida livre de progressão (Tabela 4), ou seja, a combinação de capecitabina e cisplatina foi equivalente à combinação de 5-FU e cisplatina no que se refere à mediana de sobrevida global.
Tabela 4. Resumo dos resultados dos principais parâmetros de eficácia (PPP, Estudo ML17032)
* Efeito do tratamento não ajustado em modelo proporcional de Cox.
Um estudo clínico multicêntrico, randomizado, de fase III, comparando capecitabina com 5-FU e oxaliplatina com cisplatina em pacientes com câncer gástrico avançado foi conduzido para o tratamento de primeira linha de câncer gástrico avançado. Nesse estudo, 1.002 pacientes foram randomizados em um desenho fatorial 2x2 para um dos quatro braços seguintes:
ECF
Epirrubicina (50 mg/m2 em bolus no dia 1, a cada três semanas), cisplatina (60 mg/m2, por infusão de duas horas, no dia 1, a cada três semanas) e 5-FU (200 mg/m2 por dia, administrado por infusão contínua por meio de um acesso central).
ECX
Epirrubicina (50 mg/m2 em bolus, no dia 1, a cada três semanas), cisplatina (60 mg/m2 por infusão de duas horas no dia 1, a cada três semanas) e capecitabina (625 mg/m2, duas vezes por dia, continuamente).
EOF
Epirrubicina (50 mg/m2 em bolus no dia 1, a cada três semanas), oxaliplatina (130 mg/m2 administrado em infusão de duas horas no dia 1, a cada três semanas) e 5-FU (200 mg/m2 diariamente administrado por infusão contínua por meio de acesso central).
EOX
Epirrubicina (50 mg/m2 em bolus no dia 1, a cada três semanas), oxaliplatina (130 mg/m2 administrado em infusão de duas horas no dia 1, a cada três semanas) e capecitabina (625 mg/m2, duas vezes ao dia, continuamente).
As análises de eficácia primária na população per-protocolo demonstraram a não inferioridade na sobrevida global para a capecitabina versus esquemas baseados no 5-FU (razão de risco 0,86, IC 95%: 0,80 a 0,99) e para a oxaliplatina versus esquemas baseados na cisplatina (razão de risco 0,92, IC 95%: 0,8 a 1,1).
A sobrevida média global foi de 10,9 meses em esquemas baseados na capecitabina e 9,6 meses em esquemas com 5-FU. A sobrevida média global foi de 10,0 meses em esquemas baseados em cisplatina e 10,4 meses em esquemas baseados em oxaliplatina.
Capecitabina também tem sido usada em combinação com oxaliplatina no tratamento do câncer gástrico avançado.
Câncer colorretal e gástrico avançado: meta-análise
Uma meta-análise de seis estudos clínicos (SO14695, SO14796, M66001, NO16966, NO16967, M17032) avaliou se capecitabina pode substituir 5-FU no câncer gastrintestinal. A análise conjunta inclui 3.097 pacientes tratados com esquemas que continham capecitabina e 3.074 pacientes tratados com esquemas que continham 5-FU.
A razão de risco para a sobrevida global foi de 0,94 (IC de 95%: 0,89 a 1,00, p = 0,0489), indicando que os esquemas que continham capecitabina são não inferiores aos esquemas que continham 5-FU.
Câncer de mama
Terapia combinada
Capecitabina para o tratamento de pacientes com câncer de mama metastático ou localmente avançado, em combinação com docetaxel, após insucesso de quimioterapia citotóxica incluindo uma antraciclina foi estudada em um estudo clínico multicêntrico, randomizado, controlado, de fase III.
Nesse estudo, 255 pacientes foram randomizados para tratamento com capecitabina (1.250 mg/m2, duas vezes ao dia, durante duas semanas, seguido por período de descanso de uma semana) e docetaxel (75 mg/m2, por infusão intravenosa de uma hora, a cada três semanas). Um total de 256 pacientes foram randomizados para tratamento com docetaxel isoladamente (100 mg/m2, por infusão intravenosa de uma hora, a cada três semanas).
A sobrevida foi superior no grupo tratado com a combinação capecitabina + docetaxel (p = 0,0126). A sobrevida mediana foi de 442 dias (capecitabina + docetaxel) versus 352 dias (docetaxel isoladamente). Os índices de resposta objetiva global em toda a população randomizada (avaliação do pesquisador) foram 41,6% (capecitabina + docetaxel) versus 29,7% (docetaxel isolado); p = 0,0058.
O tempo até a progressão da doença ou morte foi superior no grupo tratado com a combinação capecitabina + docetaxel (p< 0,0001). O tempo mediano até a progressão da doença foi de 186 dias (capecitabina + docetaxel) versus 128 dias (docetaxel isoladamente).
Monoterapia
Dois estudos clínicos multicêntricos, de fase II foram conduzidos para determinar o uso de capecitabina em monoterapia para o tratamento de pacientes com câncer de mama metastático ou localmente avançado após insucesso com um taxano e de regime quimioterápico com antraciclina ou para aqueles pacientes nos quais a terapia adicional com antraciclina não está indicada.
Nesses estudos, um total de 236 pacientes foi tratado com capecitabina (1.250 mg/m2, duas vezes ao dia, durante duas semanas, seguido por período de descanso de uma semana). Os índices de resposta objetiva global (avaliação do pesquisador) foram 20% (primeiro estudo) e 25% (segundo estudo). O tempo mediano até a progressão da doença foi de 93 e de 98 dias. A sobrevida mediana foi de 384 e de 373 dias.
Características farmacológicas
Farmacodinâmica
Mecanismo de ação
A capecitabina é derivada do carbamato de fluoropirimidina, um agente citotóxico tumor ativado e tumor seletivo, que foi planejado para administração oral. A capecitabina é atóxica in vitro; in vivo, no entanto, é sequencialmente convertida para a fração citotóxica 5-fluoruracila (5-FU), que, por sua vez, é posteriormente metabolizada.
A formação de 5-FU ocorre preferencialmente no tumor por um fator angiogênico associado ao tumor, denominado timidina fosforilase (dThdPase), minimizando assim a exposição dos tecidos sadios do organismo a 5-FU sistêmica.
A biotransformação enzimática sequencial da capecitabina para 5-FU leva a maiores concentrações de 5-FU nos tecidos tumorais. Após a administração oral de capecitabina para pacientes com câncer colorretal (n = 8), a razão entre a concentração de 5-FU nos tumores colorretais versus tecidos adjacentes foi de 3,2 (variação de 0,9 a 8,0).
A razão entre a concentração de 5-FU no tumor versus plasma foi de 21,4 (variação de 3,9 a 59,9), enquanto a razão entre os tecidos saudáveis e o plasma foi de 8,9 (variação de 3,0 a 25,8).
A atividade da timidina fosforilase foi quatro vezes maior no tumor colorretal primário do que no tecido adjacente normal. Diversos tumores humanos, como câncer de mama, gástrico, colorretal, colo de útero e ovariano, apresentam nível de timidina fosforilase maior [capaz de converter 5'-DFUR (5'-desoxi-5-fluorouridina) em 5-FU] do que os tecidos normais correspondentes.
Tanto as células normais quanto as células tumorais metabolizam o 5-FU para monofosfato de 5-fluoro-2-desoxiuridina (FdUMP) e trifosfato de 5-fluoruridina (FUTP). Esses metabólitos causam dano à célula por meio de dois mecanismos diferentes. Inicialmente, o FdUMP e o cofator folato N5-10-metileno-tetrahidrofolato ligam-se à timidilato sintetase (TS) para formar um complexo ternário covalente.
Essa ligação inibe a formação de timidilato a partir do uracil. A timidilato é a precursora necessária do trifosfato de timidina, que por sua vez, é essencial para a síntese de DNA, de forma que uma deficiência desse composto pode inibir a divisão celular. Além disso, as enzimas nucleares de transcrição podem incorporar FUTP erroneamente, no lugar do trifosfato de uridina (UTP), durante a síntese de RNA. Esse erro metabólico pode interferir com o processamento do RNA e com a síntese proteica.
Farmacocinética
Absorção
Após administração oral, a capecitabina é rápida e extensamente absorvida, seguida da extensa conversão nos metabólitos, 5’- desoxi-5-fluorcitidina (5’-DFCR) e 5'-desoxi-5-fluorouridina (5’-DFUR).
A administração com alimentos diminui a taxa de absorção da capecitabina, porém, com mínimo efeito sobre as áreas sob a curva (AUC) do 5’-DFUR e de seu metabólito subsequente, 5-FU.
No décimo quarto dia de administração, após a ingestão de alimento, com a dose de 1.250 mg/m2, as concentrações plasmáticas máximas (Cmáx em ?g/mL) para a capecitabina, 5’-DFCR, 5’DFUR, 5-FU e FBAL (?-fluoro-?-alanina, metabólito inativo do 5-FU) foram 4,47, 3,05, 12,1, 0,95 e 5,46, respectivamente.
Os tempos para atingir as concentrações plasmáticas máximas (Tmáx em horas) foram 1,50, 2,00, 2,00, 2,00 e 3,34. Os valores da AUC0-? em ?g*h/mL foram 7,75, 7,24, 24,6, 2,03 e 36,3.
Distribuição
Ligação proteica
Estudos realizados com plasma humano in vitro determinaram que as ligações da capecitabina, 5’-DFCR, 5’-DFUR e 5-FU às proteínas, principalmente à albumina, foram de 54%, 10%, 62% e 10%, respectivamente.
Metabolismo
A capecitabina é metabolizada inicialmente, por meio da carboxilesterase hepática, para 5’-DFCR, que é convertida a seguir em 5’- DFUR pela citidina desaminase, localizada principalmente no fígado e nos tecidos tumorais.
A formação de 5-FU ocorre preferencialmente no tumor pelo fator angiogênico associado ao tumor, dThdPase (timidina-fosforilase), minimizando assim a exposição sistêmica dos tecidos sadios do organismo a 5-FU sistêmica.
A AUC plasmática de 5-FU é 6 a 22 vezes menor que aquela observada após a administração intravenosa em bolus de 5-FU (dose de 600 mg/m2). Os metabólitos da capecitabina tornam-se citotóxicos somente após sua conversão para 5-FU e para anabólitos de 5- FU.
O 5-FU é então catabolizado, dando origem aos metabólitos inativos diidro-5-fluoruracila (FUH2), ácido 5-fluoro-ureidopropiônico (FUPA) e ?-fluoro-?-alanina (FBAL), via diidropirimidina desidrogenase (DPD), a qual é limitante da taxa.
Eliminação
As meias-vidas de eliminação (t1?2 em horas) da capecitabina, 5’-DFCR, 5’-DFUR, 5-FU e FBAL foram 0,85, 1,11, 0,66, 0,76 e 3,23, respectivamente. A farmacocinética da capecitabina foi avaliada em uma faixa de dose de 502 a 3.514 mg/m2/dia.
Os parâmetros da capecitabina, do 5’-DFCR e 5’-DFUR medidos nos dias 1 e 14 foram similares. A AUC de 5-FU foi de 30% a 35% maior no dia 14, mas não aumentou nos dias seguintes (dia 22). Com doses terapêuticas, a farmacocinética da capecitabina e de seus metabólitos foi proporcional à dose, exceto para 5-FU.
Após administração oral, os metabólitos da capecitabina são recuperados principalmente na urina, cerca de 95,5% da dose. A excreção fecal é mínima (2,6%). O principal metabólito excretado na urina é FBAL, que representa 57% da dose administrada. Aproximadamente 3% da dose administrada é excretada na urina como fármaco inalterado.
Terapia combinada
Em estudos fase I, não foram demonstrados efeitos de capecitabina sobre a farmacocinética do docetaxel ou do paclitaxel (Cmáx e AUC), assim como nenhum efeito foi observado do docetaxel ou do paclitaxel sobre a farmacocinética do 5’-DFUR (o metabólito mais importante da capecitabina).
Farmacocinética em populações especiais
A análise farmacocinética da população foi realizada após o tratamento de 505 pacientes, portadores de câncer colorretal, com capecitabina, na dose de 1.250 mg/m2, duas vezes ao dia.
Sexo, presença ou ausência de metástases hepáticas ao diagnóstico, Desempenho de Karnofsky, bilirrubina total, albumina sérica, TGO e TGP não tiveram efeito estatisticamente significativo sobre a farmacocinética do 5’-DFUR, 5-FU e FBAL.
Pacientes com insuficiência hepática em decorrência de metástases hepáticas
Nenhum efeito clinicamente significativo foi observado na bioativação e farmacocinética da capecitabina em pacientes portadores de câncer com insuficiência hepática leve a moderada devida a metástases hepáticas.
Não há dados farmacocinéticos de pacientes com insuficiência hepática grave.
Pacientes com insuficiência renal
Com base em estudo farmacocinético em pacientes com câncer e insuficiência renal leve a grave, não há evidência de efeito da depuração de creatinina sobre a farmacocinética da droga intacta e de 5-FU.
Foi constatada que a depuração de creatinina influencia a exposição sistêmica ao 5’-DFUR (35% de aumento da AUC, quando a depuração de creatinina diminui em 50%) e ao FBAL (aumento de 114% da AUC, quando a depuração de creatinina diminui em 50%). O FBAL é um metabólito sem atividade antiproliferativa; o 5’- DFUR é um precursor direto do 5-FU.
Idosos
Com base na análise farmacocinética da população, incluindo pacientes com idades de 27 a 86 anos, sendo 234 (46%) pacientes com idade superior ou igual a 65 anos, a idade não teve influência na farmacocinética do 5’-DFUR e 5-FU. A AUC do FBAL aumentou com a idade (20% no aumento da idade resultou em 15% de aumento na AUC do FBAL). Esse aumento provavelmente é devido à alteração na função renal.
Etnia
Baseado na análise farmacocinética da população, que incluiu 455 pacientes brancos (90,1%), 22 pacientes negros (4,4%) e 28 pacientes de outra raça ou etnia (5,5%), não houve diferenças quanto à farmacocinética da capecitabina entre pacientes negros e brancos.
Cuidados de Armazenamento
Xeloda deve ser conservado em temperatura ambiente (entre 15 e 30ºC).
Número de lote e datas de fabricação e validade: Vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado.
Utilize o sistema de coleta local estabelecido, se disponível.
Característica física:
Xeloda 150 mg
Cor pêssego claro.
Xeloda 500 mg
Cor pêssego.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS – 1.0100.0549
Farm. Resp.:
Tatiana Tsiomis Díaz – CRF-RJ n° 6942
Fabricado para
F. Hoffmann-La Roche Ltd., Basileia, Suíça,
por Productos Roche S.A. de C.V., Toluca, México
Embalado por:
F. Hoffmann-La Roche, Ltd., Kaiseraugst, Suíça
Registrado, importado e distribuído no Brasil por
Produtos Roche Químicos e Farmacêuticos S.A.
Est. dos Bandeirantes, 2.020 CEP 22775-109 – Rio de Janeiro – RJ
CNPJ: 33.009.945/0023-39
Serviço Gratuito de Informações – 0800 7720 289
www.roche.com.br
Venda sob prescrição médica.