para o que é indicado e para que serve?
Para que serve Xtandi é usado para tratar homens adultos com câncer de próstata que tenha se espalhado para outras partes do corpo.Continue lendo...
ofertas de
Xtandi 40Mg C 120 Cápsula...
ofertas de Xtandi 40Mg C 120 Cápsula...
R$ 12.900,00
R$ 13.071,23
R$ 13.458,70
R$ 13.458,70
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Xtandi é usado para tratar homens adultos com câncer de próstata que tenha se espalhado para outras partes do corpo.
Xtandi é um medicamento usado para tratar homens com câncer de próstata metastático resistente à castração (um tipo de câncer de próstata que é resistente a tratamentos médicos ou cirúrgicos que reduzem os níveis de testosterona do organismo e que já tenha se espalhado para outras partes do corpo) e que já tenham recebido tratamento comdocetaxel.
Xtandi não foi avaliado em crianças.
Como Xtandi funciona?
Xtandi é um medicamento que bloqueia a atividade de andrógenos (como a testosterona). Desta maneira, ele pode diminuir o crescimento do câncer de próstata e pode causar a morte das células cancerígenas e a regressão do tumor. Quando administrado diariamente, a concentração de equilíbrio de Xtandi é atingida em cerca de um mês.
Contraindicação
Xtandi é contraindicado se você
- For alérgico (hipersensível) à enzalutamida ou a qualquer dos outros ingredientes do mesmo;
- Estiver grávida ou na iminência de engravidar.
Este medicamento é contraindicado para menores de 18 anos.
Como usar
Engula as cápsulas inteiras com água.
Não mastigue, dissolva ou abra as cápsulas antes de engolir.
Deve-se evitar o uso de alguns medicamentos conhecidos como inibidores fortes do CYP2C8 ao mesmo tempo que Xtandi. Se estiver tomando um inibidor forte do CYP2C8, você deve tomar uma dose reduzida de Xtandi.
Converse com seu médico sobre os medicamentos que estiver usando, já que ajustes de dose podem ser necessários.
Posologia
Use Xtandi exatamente como prescrito.
Seu médico prescreverá a dose correta para você. A dose recomendada é 160 mg (quatro cápsulas de 40 mg), tomadas na mesma hora, uma vez ao dia, com ou sem alimentos.
Duração do tratamento
A duração média do tratamento com Xtandi em estudos clínicos foi de cerca de 12,8 meses. A duração do tratamento pode variar dependendo da condição clínica do paciente.
Este medicamento não deve ser partido, aberto ou mastigado
Siga a orientação do seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Xtandi?
Se você esquecer de tomar a dose de Xtandi no horário habitual, tome-a assim que se lembrar.
Se você se esquecer de tomar a dose de Xtandi por um dia inteiro, tome-a no dia seguinte, no horário habitual.
Se você se esquecer de tomar a dose de Xtandi por mais de um dia, fale imediatamente com seu médico.
Não tome uma dose dobrada para compensar a dose que você esqueceu.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico ou cirurgião-dentista.
Precauções
Antes de começar a usar o Xtandi, informe seu médico ou outro profissional de saúde sobre todas as suas condições clínicas, inclusive se você
- Está tomando qualquer medicamento para prevenir coágulos sanguíneos (por exemplo, varfarina, acenocumarol);
- Tem problemas de fígado;
- Tem problemas renais.
Convulsões
Na administração do Xtandi, deve-se ter cautela em pacientes com histórico de convulsões ou outros fatores predisponentes incluindo lesões cerebrais, derrame, tumores cerebrais primários ou metástases cerebrais, ou alcoolismo. Além disso, o risco de convulsões pode ser aumentado em pacientes fazendo uso concomitante de medicamentos que reduzem o limiar convulsivo.
Síndrome de Encefalopatia Posterior Reversível (PRES)
Houve relatos raros de PRES, uma condição rara e reversível que envolve o cérebro, em pacientes tratados com Xtandi. Se você tiver convulsão, piora de dor de cabeça, confusão, cegueira ou outros problemas de visão, contate seu médico o mais rápido possível.
Fertilidade, gravidez e gestação
Xtandi não é indicado para uso em mulheres.
Este medicamento pode causar danos ao embrião/feto ou possível perda gestacional se usado por mulheres que estão grávidas. Ele não deve ser tomado por mulheres grávidas, que possam ficar grávidas durante o tratamento ou que estejam amamentando.
Se você estiver mantendo relações sexuais com uma mulher que pode engravidar, use preservativo e outro método eficaz de controle da natalidade durante e por 3 meses após o tratamento com este medicamento.
Se você mantiver relações sexuais com uma mulher grávida, use um preservativo para proteger o feto.
Este medicamento possivelmente pode afetar a fertilidade masculina.
Xtandi não deve ser usado por menores de 18 anos de idade.
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Outros medicamentos e Xtandi
Informe ao seu médico se você estiver usando, tiver usado recentemente ou tiver possibilidade de usar qualquer outro medicamento. Você precisa saber os nomes dos medicamentos que estiver tomando.
Mantenha uma lista dos medicamentos com você e mostre ao seu médico quando ele prescrever um novo medicamento. Você não deve iniciar ou interromper nenhum medicamento antes de conversar com o médico que prescreveu Xtandi.
Dirigir e usar máquinas
Devido ao risco de convulsão associado com o uso do Xtandi, os pacientes devem ser informados do risco de dirigir ou usar quaisquer ferramentas ou máquinas nas quais a súbita perda de consciência possa causar danos graves para eles ou outras pessoas.
Este medicamento contém sorbitol (um tipo de açúcar).
Se você foi informado pelo seu médico que tem intolerância a alguns açúcares, contate seu médico ou outro profissional de saúde antes de usá-lo.
Informar ao seu medido ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Isto pode ser prejudicial para sua saúde.
Reações Adversas
Como todos os medicamentos, este pode causar efeitos colaterais, embora não ocorra em todo mundo.
Convulsões
Foram relatadas convulsões em 5 em cada 1.000 pessoas usando Xtandi e em menos de 1 em cada 1.000 pessoas usando placebo.
A probabilidade de convulsões é maior se você tomar uma dose maior que a recomendada desse medicamento, se estiver tomando alguns outros medicamentos ou se o seu risco de convulsão for maior que o habitual.
Você deve evitar atividades onde a perda de consciência repentina possa causar sérios danos a você ou à outras pessoas. Contate imediatamente seu médico caso você venha a ter perda de consciência ou convulsão.
Não tome mais nenhum comprimido de Xtandi.
Síndrome de Encefalopatia Posterior Reversível(PRES)
Houve relatos raros de PRES (pode afetar até 1 em 1.000 pessoas), uma condição rara e reversível que envolve o cérebro, em pacientes tratados com Xtandi. Se você tiver convulsão, piora de dor de cabeça, confusão, cegueira ou outros problemas de visão, contate seu médico o mais rápido possível.
Outros possíveis efeitos colaterais
Composição
Cada cápsula gelatinosa mole contém:
40 mg de enzalutamida.
Excipientes: Caprilcaproil macrogol glicerídeos, butil-hidroxianisol (E320), butil- hidroxitolueno (E321), gelatina, sorbitol, solução de sorbitano, glicerol, dióxido de titânio (E171), água purificada, tinta farmacêutica (etanol, acetato de etila, propilenoglicol, óxido de ferro (E172), acetato ftalato de polivinila, álcool isopropílico, macrogol 400, solução de amônia).
Superdosagem
Se você tomar mais cápsulas do que o prescrito, suspenda o uso do Xtandi e contate seu médico. Você pode ter um risco aumentado de convulsão ou outros efeitos colaterais.
Vá a um serviço de emergência, levando com você a caixa e a bula deste produto.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Potencial para outros medicamentos afetarem exposições a Enzalutamida (substância ativa)
Potencial para Enzalutamida (substância ativa) em afetar exposições a outros medicamentos
Os grupos de medicamentos que podem ser afetados incluem os seguintes medicamentos, mas não estão limitados a eles
Certos medicamentos usados para tratar asma e outras doenças respiratórias (ex.: aminofilina, teofilina).
Medicamentos usados para tratar certos transtornos psiquiátricos como depressão e esquizofrenia (ex.: clozapina, olanzapina, risperidona, ziprasidona, bupropiona, lítio, clorpromazina, mesoridazina, tioridazina, amitriptilina, desipramina, doxepina, imipramina, maprotilina, mirtazapina).
- Analgésicos (ex.: fentanila, tramadol, petidina);
- Antibióticos (ex.: claritromicina, doxiciclina);
- Agentes anticâncer (ex.: cabazitaxel);
- Anticoagulantes (ex.: acenocumarol, varfarina);
- Antiepiléticos (ex.: carbamazepina, clonazepam, fenitoína, primidona, ácido valproico);
- Antipsicóticos (ex.: haloperidol);
- Betabloqueadores (ex.: bisoprolol, propranolol);
- Benzodiazepínicos (ex.: diazepam, midazolam, zolpidem);
- Bloqueadores dos canais de cálcio (ex.: diltiazem, felodipina, nicardipina, nifedipina, verapamil);
- Glicosídeos cardíacos (ex.: digoxina);
- Corticoides (ex.: dexametasona, prednisolona);
- Antivirais HIV (ex.: indinavir, ritonavir);
- Estatinas metabolizadas pelo CYP3A4 (ex.: atorvastatina, sinvastatina);
- Agentes tiroideanos (ex.: levotiroxina).
O potencial total de indução da Enzalutamida (substância ativa) pode não ocorrer até cerca de 1 mês após o início do tratamento, quando as concentrações plasmáticas da Enzalutamida (substância ativa) no estado de equilíbrio forem atingidas, embora alguns efeitos de indução possam aparecer anteriormente.
Os pacientes em uso de medicamentos que são substratos do CYP2B6, CYP3A4, CYP2C9, CYP2C19, CYP1A2 ou UGT1A1 devem ser avaliados para possível perda de efeitos farmacológicos (ou aumento dos efeitos nos casos em que há formação de metabólitos ativos) durante o primeiro mês de tratamento com Enzalutamida (substância ativa), e o ajuste de dose deve ser considerado conforme apropriado. Levando em consideração a longa meia-vida da Enzalutamida (substância ativa), o efeito nas enzimas pode persistir por um mês ou mais após a interrupção da Enzalutamida (substância ativa).
Uma redução gradual da dose do medicamento concomitante pode ser necessária quando ao interromper o tratamento com Enzalutamida (substância ativa).
Interação Alimentícia
Os alimentos não têm efeito clinicamente significativo sobre o grau de exposição à Enzalutamida (substância ativa). Em ensaios clínicos, foi administrada Enzalutamida (substância ativa) independentemente dos alimentos.
Ação da Substância
Resultados da eficácia
Eficácia clínica e segurança
A eficácia da Enzalutamida (substância ativa) foi estabelecida em dois estudos clínicos de Fase 3, multicêntricos, randomizados e controlados por placebo [CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] de pacientes com câncer de próstata metastático e progressivo que não responderam à terapia de privação de andrógeno [análogo do hormônio liberador do hormônio luteinizante (LHRH) ou após orquiectomia bilateral].
O estudo PREVAIL incluiu pacientes não tratados previamente com quimioterapia, enquanto o estudo AFFIRM incluiu pacientes que já haviam recebido docetaxel anteriormente. Todos os pacientes continuaram com o análogo de LHRH ou realizaram orquiectomia bilateral prévia.
Enzalutamida (substância ativa) foi administrada por via oral na dose diária de 160 mg no grupo de tratamento ativo.
Em ambos os estudos clínicos, os pacientes do grupo controle receberam placebo e foram autorizados a tomar prednisona (a dose máxima diária permitida foi de 10 mg de prednisona ou outro equivalente), embora não fosse obrigatório. Alterações da concentração sanguínea de PSA nem sempre indicam benefício clínico de forma independente. Portanto, em ambos os estudos recomendou-se que os pacientes mantivessem seus tratamentos do estudo até que os critérios de descontinuação fossem atingidos, conforme especificado abaixo para cada um dos estudos.
Estudo MDV3100-03 (PREVAIL) (pacientes não tratados previamente com quimioterapia)
Foi randomizado, em distribuição de 1:1, um total de 1.717 pacientes assintomáticos ou ligeiramente sintomáticos, não tratados previamente com quimioterapia, para receber Enzalutamida (substância ativa) por via oral na dose de 160 mg, uma vez ao dia (N = 872), ou placebo por via oral, uma vez ao dia (N = 845).
Foi permitida a inclusão de pacientes com doença visceral, pacientes com histórico de insuficiência cardíaca leve a moderada (NYHA Classes 1 ou 2) e pacientes em uso de medicações associadas à diminuição do limiar convulsivo. Foi administrada pelo menos uma medicação concomitante conhecida por diminuir o limiar convulsivo em 52,5% dos pacientes no grupo de tratamento com Enzalutamida (substância ativa), em comparação com 45,9% dos pacientes do grupo tratado com placebo.
Foram excluídos pacientes com histórico de convulsão ou alguma condição que pudesse predispor à convulsão e pacientes com dor moderada ou grave decorrente do câncer de próstata. O tratamento do estudo continuou até a progressão da doença (evidência de progressão radiográfica, evento relacionado ao esqueleto ou progressão clínica) e o início de quimioterapia citotóxica ou de algum agente em investigação, ou até toxicidade inaceitável.
Os dados demográficos dos pacientes e as características basais da doença foram equilibrados entre os grupos de tratamento. A idade mediana foi 71 anos (variação 42-93) e a distribuição racial foi de 77% caucasianos; 10% asiáticos; 2% negros e 11% outras raças ou desconhecido. Sessenta e oito por cento dos pacientes apresentavam índice de desempenho ECOG 0 e 32% dos pacientes apresentavam ECOG 1.
A avaliação basal da dor foi de 0-1 (assintomático) em 67% dos pacientes e de 2-3 (ligeiramente sintomático) em 32% dos pacientes, conforme definido pelo Inventário Breve de Dor - Forma Abreviada (pior dor relatada pelo paciente ao longo das 24 horas anteriores, em escala de 0 a 10). Aproximadamente 45% dos pacientes apresentaram doença mensurável nos tecidos moles ao ingressar no estudo, e 12% dos pacientes tinham metástases viscerais (pulmão e/ou fígado).
Os desfechos coprimários de eficácia foram a sobrevida global e a sobrevida livre de progressão radiográfica (rPFS). Além dos desfechos coprimários, o benefício também foi avaliado usando o tempo até o início de quimioterapia citotóxica, melhor resposta global nos tecidos moles, tempo até o primeiro evento relacionado ao esqueleto, resposta do PSA (diminuição ? 50% em relação ao valor basal), tempo até progressão do PSA e tempo até a degradação da pontuação total do questionário de qualidade de vida FACT-P (Avaliação Funcional de Terapia de Câncer – Próstata).
A progressão radiográfica foi avaliada com uso de estudos de imagens sequenciais de acordo com os critérios definidos pelo Grupo de Trabalho dos Ensaios Clínicos de Câncer da Próstata 2 (PCWG2 - Prostate Cancer Clinical Trials Working Group 2) (para lesões ósseas) e/ou com os critérios dos Critérios de Avaliação de Resposta em Tumores Sólidos (Response Evaluation Criteria in Solid Tumors - RECIST v 1.1) (para lesões em tecidos moles). A análise da rPFS utilizou revisões centrais das avaliações radiográficas da progressão.
Durante a análise interina pré-especificada para sobrevida global, o tratamento com Enzalutamida (substância ativa) demonstrou melhora estatisticamente significativa na sobrevida global comparada com o placebo, com redução do risco de morte em 29,4% [RR = 0,706; (IC 95%: 0,596; 0,837), p < 0,0001]. No momento da análise interina, haviam falecido 27,6% (241 de 872) dos pacientes tratados com Enzalutamida (substância ativa), em comparação com 35,4% (299 de 845) dos pacientes tratados com placebo.
A sobrevida global mediana estimada foi de 32,4 meses (IC 95%: 30,1, não alcançada) nos pacientes tratados com Enzalutamida (substância ativa) e 30,2 meses (IC 95%: 28,0, não alcançada) nos pacientes com placebo (Tabela 1 e Figuras 1 e 2). Além disso, 40,3% dos pacientes tratados com Enzalutamida (substância ativa) e 70,6% dos pacientes tratados com placebo receberam terapias subsequentes com demonstração de benefício quanto à sobrevida.
Os resultados de uma análise de sobrevida global exploratória atualizada realizada antes do cruzamento dos pacientes para o tratamento aberto com Enzalutamida (substância ativa), que incluíram 116 eventos adicionais, foram consistentes com a análise interina (Tabela 1 e Figura 1).
Tabela 1: Sobrevida Global de Pacientes Tratados com Enzalutamida (substância ativa) ou Placebo no estudo PREVAIL (Análise por Intenção de Tratar):
| Enzalutamida (substância ativa) (N = 872) | Placebo (N = 845) | |
| Análise interina pré-especificada | ||
| Número de óbitos (%) | 241 (27,6%) | 299 (35,4%) |
| Mediana, meses (IC 95%) | 32,4 (30,1; NR) | 30,2 (28,0; NR) |
| Valor-pa | < 0,0001 | |
| Razão de risco (IC 95%)b | 0,71 (0,60, 0,84) | |
a Valor-p é derivado de um teste de log-rank não estratificado.
b A razão de risco é derivada de um modelo de risco proporcional não estratificado. Uma razão de risco < 1 favorece a Enzalutamida (substância ativa). NR, não alcançado.
Figura 1: Curvas de Sobrevida Global de Kaplan-Meier no estudo PREVAIL (Análise por Intenção de Tratar):
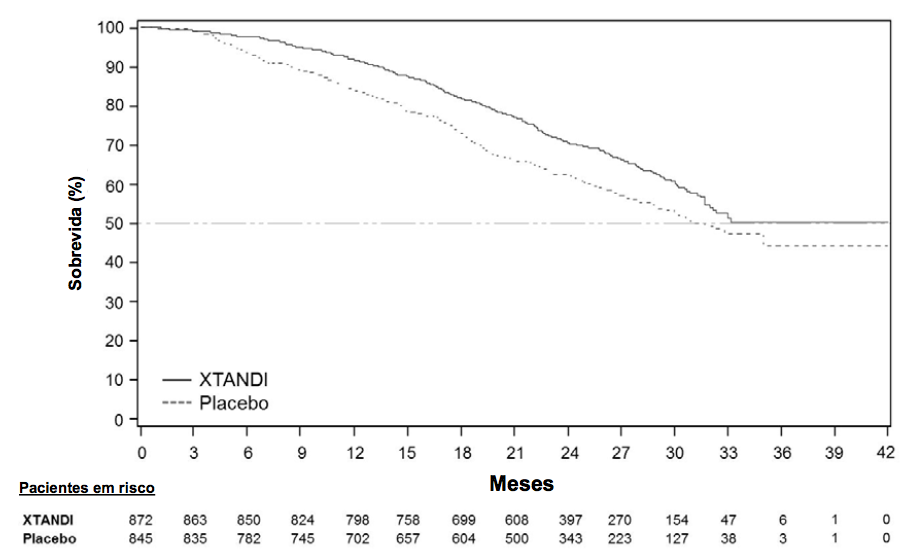
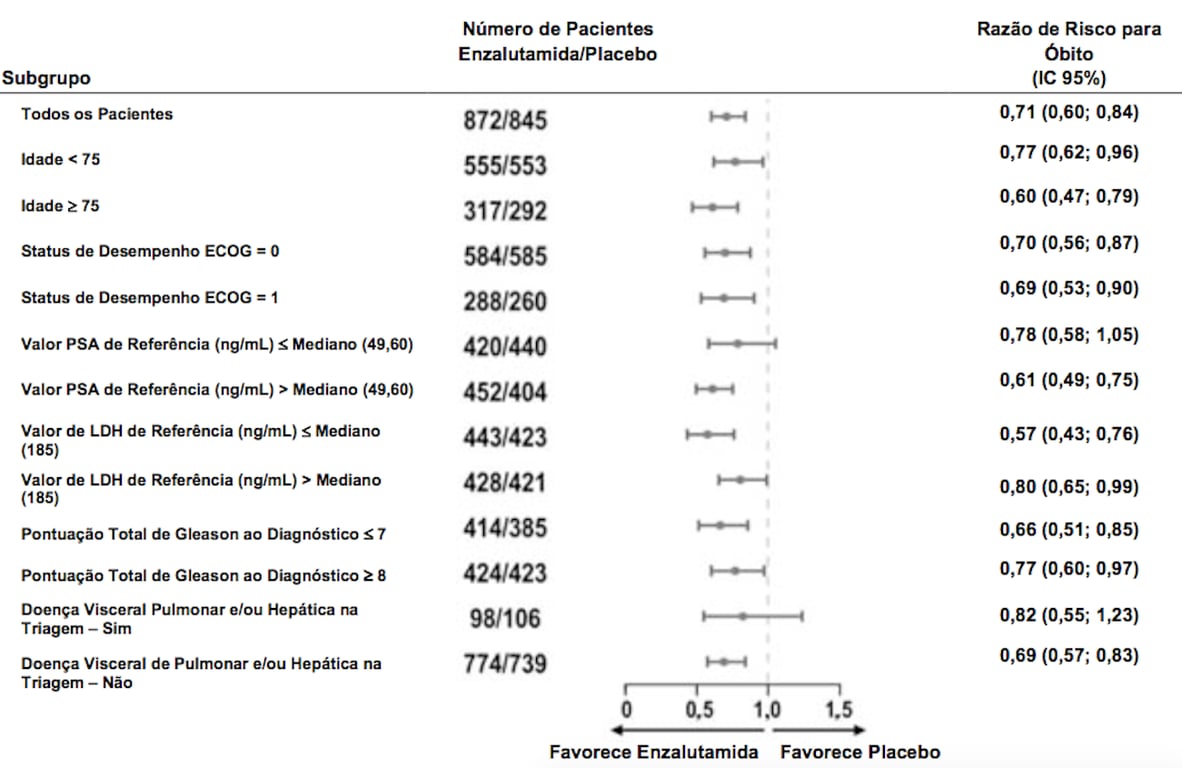
Figura 2: Sobrevida Global por Subgrupo: Razão de Risco e Intervalo de Confiança de 95% no Estudo PREVAIL (Análise por Intenção de Tratar):
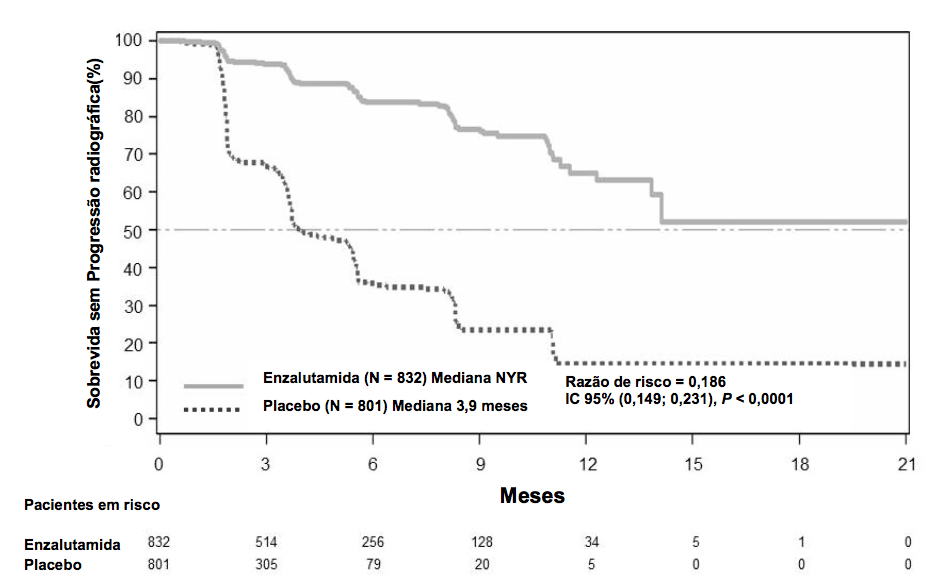
Durante a análise pré-especificada de rPFS, foi demonstrada melhora estatisticamente significativa entre os grupos de tratamento com redução de 81,4% do risco de progressão radiográfica ou morte [RR = 0,186 (IC 95%: 0,149; 0,231), p < 0,0001]. Cento e dezoito (14%) dos pacientes tratados com Enzalutamida (substância ativa) e 321 (40%) dos pacientes tratados com placebo apresentaram um evento. A mediana de rPFS não foi alcançada (IC 95%: 13,8, não alcançada) no grupo de pacientes tratados com Enzalutamida (substância ativa) e foi de 3,9 meses (IC 95%: 3,7; 5,4) no grupo de pacientes com placebo (Figuras 3).
Foi observado um benefício consistente quanto a rPFS em todos os subgrupos pré-especificados de pacientes (ex., idade, desempenho do ECOG de referência, PSA e LDH de referência, pontuação de Gleason ao diagnóstico e doença visceral na triagem). Uma análise do seguimento pré-especificado de rPFS com base na avaliação do investigador da progressão radiográfica demonstrou melhora estatisticamente significativa entre os grupos de tratamento, com redução de 69,3% no risco de progressão radiográfica ou morte [RR = 0,307 (IC 95%: 0,267; 0,353), p < 0,0001]. A rPFS mediana foi de 19,7 meses no grupo Enzalutamida (substância ativa) e de 5,4 meses no grupo placebo.
Figura 3: Curvas de Kaplan-Meier de Sobrevida Global Livre de Progressão Radiográfica no estudo PREVAIL (Análise por Intenção de Tratar):
No momento da análise primária havia 1.633 pacientes randomizados.
Além dos desfechos coprimários de eficácia, também foram demonstradas melhoras estatisticamente significativas nos seguintes desfechos prospectivamente definidos.
O tempo mediano até início da quimioterapia citotóxica foi de 28,0 meses para pacientes recebendo Enzalutamida (substância ativa) e de 10,8 meses para pacientes recebendo placebo (RR = 0,350; IC 95%: [0,303; 0,403]; p < 0,0001).
A proporção de pacientes tratados com Enzalutamida (substância ativa) com doença mensurável na avaliação basal do estudo que tiveram uma resposta objetiva dos tecidos moles foi de 58,8% (IC 95%: 53,8; 63,7) em comparação com 5,0% (IC 95%: 3,0; 7,7) dos pacientes que receberam placebo. A diferença absoluta na resposta objetiva dos tecidos moles entre Enzalutamida (substância ativa) e placebo foi de 53,9% (IC 95%: 48,5%; 59,1%; p < 0,0001).
Respostas completas foram relatadas em 19,7% dos pacientes tratados com Enzalutamida (substância ativa), em comparação com 1,0% dos pacientes tratados com placebo, e respostas parciais foram relatadas em 39,1% dos pacientes tratados com Enzalutamida (substância ativa), contra 3,9% dos pacientes tratados com placebo.
A Enzalutamida (substância ativa) diminuiu significativamente o risco do primeiro evento relacionado ao esqueleto em até 28% [RR = 0,718 (IC 95%: 0,610; 0,844), valor-p < 0,0001]. Um evento relacionado ao esqueleto foi definido como radioterapia ou cirurgia óssea para câncer de próstata, fratura patológica de osso, compressão da medula espinhal ou mudança de terapia antineoplásica para tratar a dor óssea.
A análise incluiu 587 eventos relacionados ao esqueleto, dos quais 389 eventos (66,3%) foram de radiação óssea, 79 eventos (13,5%) foram de compressão da medula espinhal, 70 eventos (11,9%) foram de fratura patológica de osso, 45 eventos (7,6%) foram de mudança de terapia antineoplásica para tratar a dor óssea, e 22 eventos (3,7%) foram de cirurgia óssea.
Os pacientes que recebem Enzalutamida (substância ativa) demonstraram uma superioridade estatisticamente significativa na taxa de resposta total do PSA (definida como uma redução ? 50% desde o basal), em comparação com os pacientes que receberam placebo, 78,0% versus 3,5% (diferença = 74,5%; p < 0,0001).
O tempo mediano para progressão do PSA de acordo com os critérios do PCWG2 foi de 11,2 meses para pacientes tratados com Enzalutamida (substância ativa) e de 2,8 meses para pacientes que receberam placebo [RR = 0,169, (IC 95%: 0,147; 0,195); p < 0,0001].
O tratamento com Enzalutamida (substância ativa) diminuiu o risco de degradação da avaliação FACT-P em 37,5% em comparação com o placebo (p < 0,001). O tempo mediano até a degradação da avaliação FACT-P foi de 11,3 meses no grupo Enzalutamida (substância ativa) e 5,6 meses no grupo de placebo.
Estudo CRPC2 (AFFIRM) (pacientes que receberam quimioterapia prévia)
A eficácia e segurança de Enzalutamida (substância ativa) em pacientes com câncer de próstata metastático resistente à castração que tenham recebido docetaxel e estavam usando um análogo do LHRH ou que foram submetidos à orquiectomia, foram avaliados por um ensaio clínico de Fase 3, multicêntrico, controlado por placebo, randomizado. Um total de 1.199 pacientes foram randomizados em proporção 2:1 para receber tanto Enzalutamida (substância ativa) oralmente, em uma dose de 160 mg uma vez ao dia (N = 800), ou placebo uma vez ao dia (N = 399).
Foi permitido, mas não exigido, que os pacientes tomassem prednisona. Os pacientes randomizados para cada unidade tinham que continuar o tratamento até a progressão da doença (definida como progressão confirmada por radiografia ou a ocorrência de um evento relacionado ao esqueleto) e iniciação de novo tratamento antineoplásico sistêmico, toxicidade inaceitável ou abstinência.
Os dados demográficos de pacientes e características de referência da doença a seguir foram equilibrados entre as unidades de tratamento. A idade média foi 69 anos (variação 41-92) e a distribuição racial foi 92,7% caucasianos; 3,9% negros; 1,1% asiáticos e 2,1% outras raças. O índice de desempenho do ECOG foi 0-1 em 91,5% dos pacientes e 2 em 8,5% dos pacientes; 28,4% tiveram um índice de Inventário Breve de Dor de ? 4 (média de pior dor relatada por paciente sobre as 24 horas anteriores calculada por 7 dias antes da randomização).
A maioria (91,2%) dos pacientes tinham metástases nos ossos e 23,2% tinham comprometimento visceral de pulmão e/ou fígado. Ao ingressar no estudo, 41% dos pacientes randomizados apresentaram apenas progressão do PSA, enquanto 59% dos pacientes apresentaram progressão radiográfica. Cinquenta e um (51%) dos pacientes estavam em uso de bisfosfonados no período basal.
A análise interina pré-especificada do protocolo após 520 óbitos demonstrou uma superioridade estatisticamente significativa na sobrevida global mediana em pacientes tratados com Enzalutamida (substância ativa) comparado com o placebo (Tabela 2 e Figura 4).
Tabela 2 Sobrevida Global de Pacientes Tratados com Enzalutamida (substância ativa) ou Placebo no estudo AFFIRM (Análise por Intenção de Tratar):
| Enzalutamida (substância ativa) (N = 800) | Placebo (N = 399) | |
| Óbitos (%) | 308 (38,5%) | 212 (53,1%) |
| Sobrevida Mediana (meses) (IC 95%) | 18,4 (17,3; NR) | 13,6 (11,3; 15,8) |
| Valor pa | < 0,0001 | |
| Razão de risco (IC 95%)b | 0,631 (0,529; 0,752) | |
a O valor p é derivado de um teste log-rank estratificado pelo status do índice de desempenho ECOG (0-1 vs. 2) e pontuação média de dor (< 4 vs. ? 4).
b Razão de Risco é derivada de um modelo de risco proporcional estratificado. Razão de risco < 1 favorece Enzalutamida (substância ativa).
Figura 4: Curvas de Sobrevida Global de Kaplan-Meier no estudo AFFIRM (Análise por Intenção de Tratar):
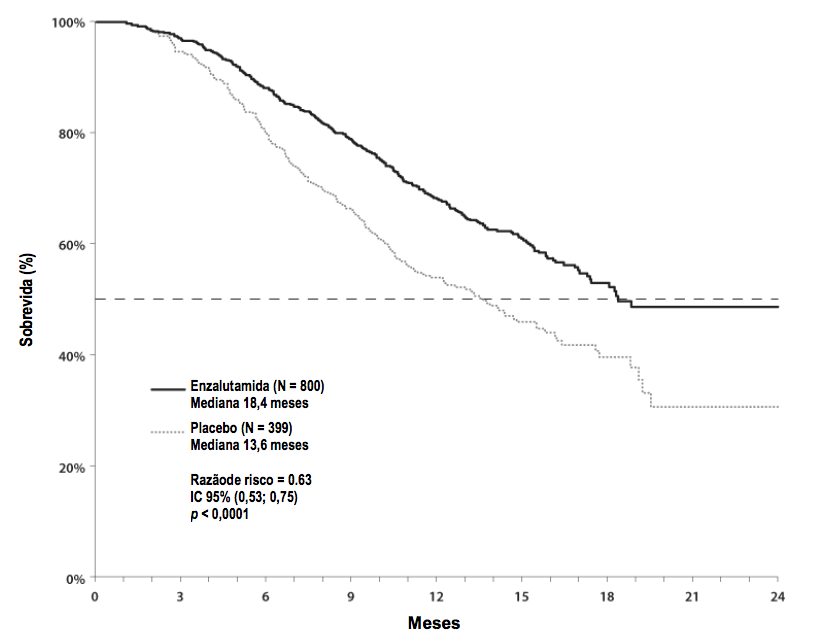
Figura 5: Sobrevida Global por Subgrupo no estudo AFFIRM – Razão de Risco e 95% de Intervalo de Confiança:
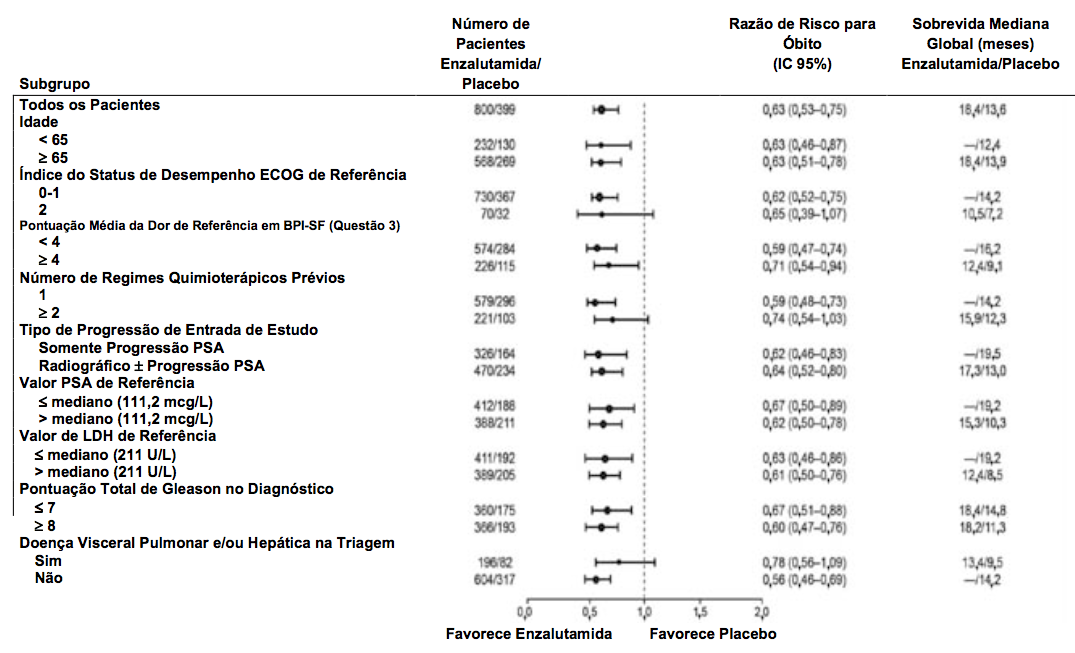
Em adição ao aumento de sobrevida global, os principais desfechos secundários (progressão do PSA, sobrevida livre de progressão radiográfica e tempo até o primeiro evento relacionado ao esqueleto) favoreceram a Enzalutamida (substância ativa) e foram estatisticamente importantes após o ajuste para testagem múltipla como segue:
A sobrevida livre de progressão radiográfica, conforme avaliada pelo investigador utilizando o RECIST v 1.1 para tecidos moles, e o aparecimento de duas ou mais lesões ósseas em exame ósseo foi de 8,3 meses para pacientes tratados com Enzalutamida (substância ativa) e de 2,9 meses para pacientes que receberam placebo (RR = 0,404; IC 95%: [0,350; 0,466]; p < 0,0001). A análise envolveu 216 óbitos sem progressão documentada e 645 eventos documentados de progressão, dos quais 303 (47%) foram devido à progressão em tecidos moles, 268 (42%) foram devido à progressão de lesão óssea e 74 (11%) foram devido à ambos, progressão em tecidos moles e lesões ósseas.
A redução de PSA confirmada de 50% ou 90% foi de 54,0% e 24,8% para pacientes tratados com Enzalutamida (substância ativa) e 1,5% e 0,9% para pacientes que receberam placebo (p < 0,0001). O tempo mediano para progressão do PSA foi de 8,3 meses para pacientes tratados com Enzalutamida (substância ativa) e de 3,0 meses para pacientes que receberam placebo (RR = 0,248; IC 95%: [0,204; 0,303]; p < 0,0001).
O tempo mediano para o primeiro evento relacionado ao esqueleto (SER) foi de 16,7 meses para pacientes tratados com Enzalutamida (substância ativa) e de 13,3 meses para pacientes que receberam placebo (RR = 0,688; IC 95%: [0,566; 0,835]; p < 0,0001). Evento relacionado ao esqueleto foi definido como uma terapia de radiação ou cirurgia óssea, fratura patológica de osso, compressão da medula espinhal ou mudança de terapia antineoplásica para tratar a dor óssea.
A análise envolveu 448 eventos relacionados ao esqueleto, dos quais 277 eventos (62%) foram de radioterapia óssea, 95 eventos (21%) foram de compressão da medula espinhal, 47 eventos (10%) foram de fratura patológica de osso, 36 eventos (8%) foram de mudança de terapia antineoplásica para tratar a dor óssea e 7 eventos (2%) foram de cirurgia óssea.
A eficácia de Enzalutamida (substância ativa) não foi estudada em pacientes que tenham recebido anteriormente o acetato de abiraterona.
A taxa de resposta para qualidade de vida (Avaliação Funcional de Terapia de Câncer – Próstata; FACT-P) foi 43,2% para pacientes tratados com Enzalutamida (substância ativa) e 18,3% para pacientes que receberam placebo (p < 0,0001).
A taxa de resposta radiográfica objetiva avaliada pelo investigador (definida como a soma das respostas total e parcial) entre pacientes tratados com Enzalutamida (substância ativa) foi 28,9% comparado com uma taxa de resposta objetiva de 3,8% para pacientes que receberam placebo (p < 0,0001).
O risco de progressão da dor foi reduzido em 44% para pacientes tratados com Enzalutamida (substância ativa) comparado aos pacientes que receberam placebo (RR = 0,56; IC 95%: [0,41; 0,78]; p = 0,0004). O tempo mediano para progressão da dor foi de 13,8 meses para pacientes que receberam placebo e não foi alcançado nos pacientes tratados com Enzalutamida (substância ativa). A progressão da dor foi definida como um aumento acima do valor de referência na avaliação FACT-P da dor, confirmada por uma segunda avaliação consecutiva obtida 3 ou mais semanas depois.
Características Farmacológicas
Mecanismo de ação
Sabe-se que o câncer de próstata é sensível à andrógenos e responde à inibição de sinalização de receptor de andrógenos (RA). Apesar dos baixos ou mesmo imperceptíveis níveis de andrógeno sérico, a sinalização RA continua a promover a progressão da doença. A estimulação do crescimento das células do tumor via receptor de andrógeno exige localização nuclear e ligação ao DNA.
A Enzalutamida (substância ativa) é um potente inibidor da sinalização do receptor de andrógenos que bloqueia vários passos no caminho da sinalização deste receptor. A Enzalutamida (substância ativa) inibe competitivamente a ligação dos andrógenos aos receptores dos mesmos, inibe a translocação nuclear de receptores ativados e inibe a associação do receptor de andrógenos ativados com o DNA mesmo no caso de superexpressão do receptor de andrógenos e nas células de câncer de próstata resistentes a antiandrógenos.
O tratamento com Enzalutamida (substância ativa) diminui o crescimento das células de câncer de próstata e pode induzir a morte das células do câncer e a regressão do tumor. Em estudos pré-clínicos, a Enzalutamida (substância ativa) carece de atividade agonista do receptor de andrógenos.
Efeitos farmacológicos
O antígeno prostático específico (PSA) funciona como marcador biológico em pacientes com câncer de próstata.
Em um estudo clínico Fase 3 de pacientes cuja quimioterapia anterior com docetaxel falhou, 54% dos pacientes tratados com Enzalutamida (substância ativa), comparado a 1,5% de pacientes que receberam placebo, tiveram pelo menos uma redução de 50% do valor de referência dos níveis de PSA.
Propriedades farmacocinéticas
A farmacocinética de Enzalutamida (substância ativa) foi avaliada em pacientes com câncer de próstata e em indivíduos saudáveis do sexo masculino. A meia-vida terminal média (t½) para Enzalutamida (substância ativa) em pacientes após uma única dose oral é 5,8 dias (variação 2,8 a 10,2 dias) e o estado de equilíbrio é atingido em aproximadamente um mês.
Com administração oral diária, a Enzalutamida (substância ativa) acumula aproximadamente 8,3 vezes relativo uma única dose. As variações diárias nas concentrações plasmáticas são baixas (taxa de máxima para mínima de 1,25). A depuração da Enzalutamida (substância ativa) ocorre principalmente via metabolismo hepático, produzindo um metabólito ativo que circula aproximadamente na mesma concentração da Enzalutamida (substância ativa).
Populações especiais
Cuidados de Armazenamento
Xtandi deve ser armazenado à temperatura ambiente (entre 15°C e 30°C). Proteja da umidade. A validade é de 24 meses após a data de fabricação.
Não use este medicamento após a data de validade exibida na embalagem. A data de validade corresponde ao último dia daquele mês.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use o medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características organolépticas
Xtandi é fornecido em cápsulas gelatinosas moles, ovais, de cor branca a quase branco, com impressão “ENZ” em tinta preta em um dos lados.
Não use qualquer cápsula que esteja vazando, danificada ou com sinais de adulteração.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderáusá-lo.
Dizeres Legais
Registro MS-1.7717.0006
Farmacêutico Responsável:
Sandra Winarski
CRF-SP: 18.496.
Fabricado por:
Catalent Pharma Solutions, LLC
St. Petersburg
FL 33716,EUA.
Embalado por:
Packaging Coordinators Inc.
Woodstock
IL 60098, EUA.
Ou
AndersonBrecon Inc.,
Rockford
IL 61109, EUA..
Registrado e importado por:
Astellas Farma Brasil Importação e Distribuição de Medicamentos Ltda.
Av. Guido Caloi, 1.935, Bloco B, 2o andar
Santo Amaro, CEP: 05802-140
São Paulo-SP
CNPJ 07.768.134/0001-04.
Xtandi é uma marca registrada da Astellas Pharma Inc.
Venda sob prescrição médica.
informações complementares
| Fabricante |
| ASTELLAS FARMA |
| Princípio ativo |
| Enzalutamida |
| Categoria do medicamento |
| Medicamentos Especiais |