para o que é indicado e para que serve?
Para que serve Eligard é indicado no tratamento do câncer de próstata avançado.Continue lendo...
ofertas de
Eligard - 22,5Mg Injetáve...
ofertas de Eligard - 22,5Mg Injetáve...
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Eligard é indicado no tratamento do câncer de próstata avançado.
Como Eligard funciona?
Eligard atua sobre o câncer de próstata avançado atenuando seus sintomas.
Contraindicação
Você não deve usar Eligard se tiver hipersensibilidade (alergia) aos hormônios estimulantes dos testículos (LH-RH), análogos agonistas (substâncias semelhantes) ao LH-RH ou a qualquer componente da formulação.
Este medicamento é contraindicado para uso por mulheres.
Este medicamento é contraindicado para uso por pacientes pediátricos.
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Este medicamento pode causar malformação ao bebê durante a gravidez.
Como usar
Eligard deve ser administrado por via subcutânea na parede abdominal (barriga), onde formará um depósito sólido de liberação lenta do medicamento.
O conteúdo da seringa é de dose única.
Assim como os demais medicamentos administrados por injeção subcutânea, o local de injeção deverá ser alterado a cada aplicação.
Eligard é preenchido e fornecido em duas seringas estéreis (livres de contaminação) separadas, cujo conteúdo deve ser misturado imediatamente antes da administração.
Importante: Deixe o produto atingir a temperatura ambiente antes de prepará-lo e utilizá-lo.
Eligard possui 2 blísteres: um identificado como diluente contendo a seringa A estéril preenchida com o diluente (sistema polimérico) Atrigel, um êmbolo branco longo reposicionável e sachê dessecante; o outro identificado como Eligard contendo a seringa B estéril preenchida com pó liófilo de acetato de leuprorrelina, agulha estéril descartável para aplicação e sachê dessecante (figura 1).
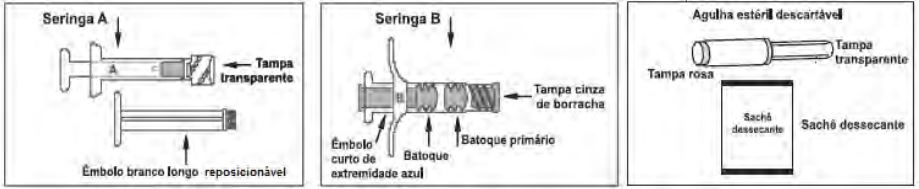
Figura 1: imagens das seringas de E.
Você deve seguir as instruções abaixo para garantir a preparação adequada de Eligard antes da administração:
- Em um local limpo, abra todas as embalagens e retire seu conteúdo. Descarte o sache dessecante.
- Puxe o êmbolo curto de extremidade azul da Seringa B. Este êmbolo curto deverá sair junto com o batoque cinza e o conjunto será descartado (Figura 2). Introduza suavemente o êmbolo branco longo reposicionável no batoque remanescente da Seringa B, girando-o no local (Figura 3).
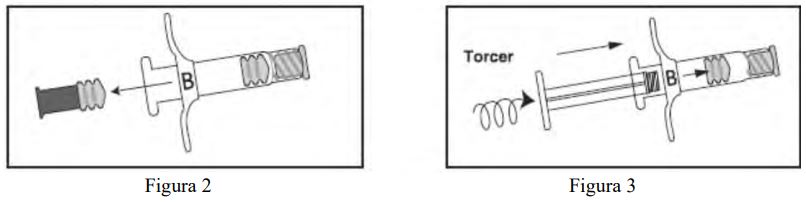
- Desrosqueie a tampa transparente da Seringa A (Figura 4). Remova a tampa cinza de borracha da Seringa B (Figura 5).
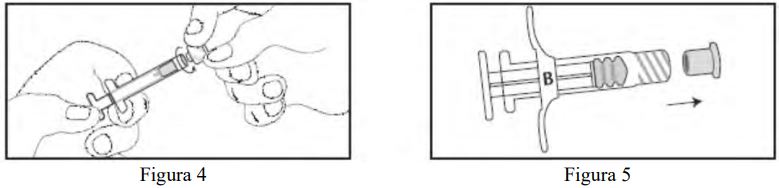
- Conecte as duas seringas, girando-as até que estejam firmemente conectadas (Figura 6).
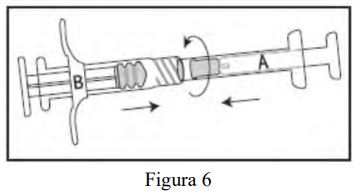
- Misture totalmente o produto, empurrando o conteúdo das seringas para frente e para trás (durante aproximadamente 45 segundos) para obter uma suspensão uniforme (Figura 7). Depois de misturada de modo uniforme, a suspensão apresentará uma coloração de amarela clara a amarela.
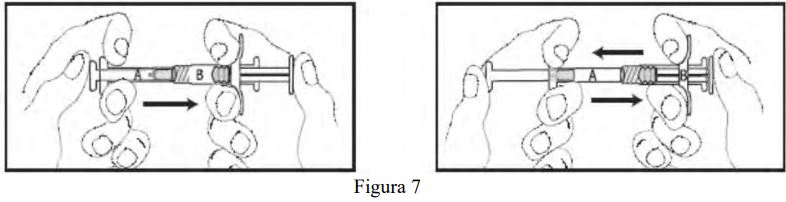
Observação: O produto deverá ser misturado conforme descrito; a agitação não fornecerá a mistura adequada do produto.
- Segure as seringas em posição vertical, com a Seringa B para baixo. As seringas deverão permanecer acopladas firmemente. Transfira todo o conteúdo do produto misturado para a Seringa B (seringa curta e larga) pressionando o êmbolo da Seringa A e puxando levemente o êmbolo da Seringa B. Continue pressionando para baixo o êmbolo da Seringa A no momento que a mesma for desconectada (Figura 8). Observação: é aceitável que pequenas bolhas de ar permaneçam na formulação.
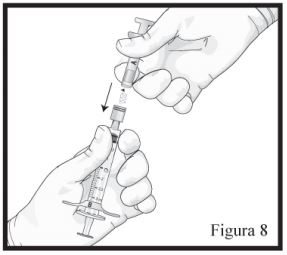
Observação: é aceitável que pequenas bolhas de ar permaneçam na formulação.
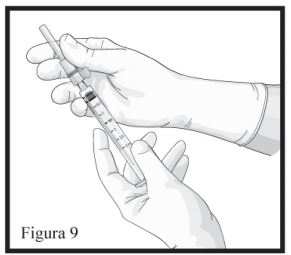
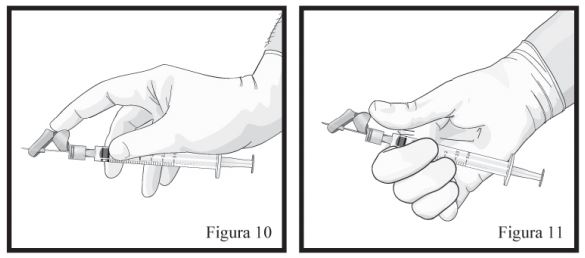
- Mantenha a Seringa B em posição vertical. Conecte a agulha à extremidade da Seringa B (Figura 9) empurrando e girando o cartucho da agulha até que esteja firmemente acoplado. Não encaixe a agulha na seringa antes de remover sua tampa rosa. Remova o capuz protetor da agulha antes da administração (Figura 09). Após a administração, não tente desconectar a agulha, trave o dispositivo de segurança: com o dedo indicador (figura 10), com o dedo polegar (figura 11), ou em uma superfície plana (figura 12). Observe a posição travada por um “click” audível e tátil. A posição travada irá cobrir completamente a agulha (Figura 13), descarte todos os componentes de modo seguro em local adequado para materiais biológicos.
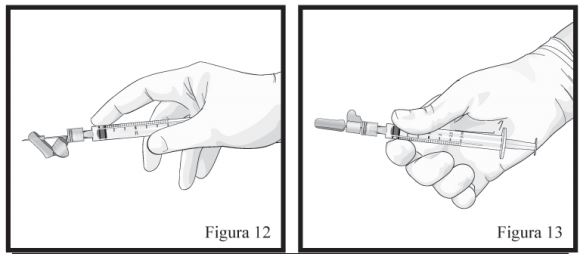
Procedimento de Administração
Importante: Deixar o produto atingir temperatura ambiente antes de utilizá-lo. Uma vez misturado, o produto deve ser administrado em até 30 minutos.
- Escolher o local da injeção no abdômen, parte superior das nádegas ou em qualquer lugar com quantidade adequada de tecido subcutâneo que não tenha pigmentação excessiva, nódulos, lesões ou pelos. Como você pode variar o local para uma injeção subcutânea, escolha uma área que não tenha sido utilizada recentemente.
- Limpe a área entorno do local da injeção com um algodão umedecido em álcool.
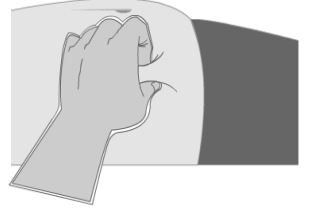
- Utilizando o polegar e o dedo indicador da sua mão não dominante, aperte a área da pele ao redor do local da injeção, formando uma prega conforme mostra a figura ao lado.
- Utilizando a sua mão dominante, inserir a agulha rapidamente em um ângulo de 90°. O ângulo aproximado que você usar dependerá da quantidade e da plenitude do tecido subcutâneo e do comprimento da agulha. Após a agulha ser inserida, solte a pele da mão não dominante.
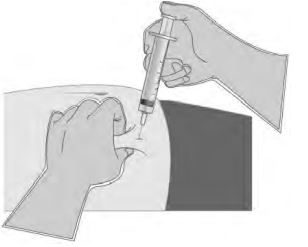
- Injetar o medicamento usando uma pressão lenta e constante. Pressione o êmbolo até a seringa ser esvaziada.
- Retirar rapidamente a agulha no mesmo ângulo utilizado para a inserção.
- Descartar todos os componentes de modo seguro em um recipiente apropriado para materiais biológicos.
Posologia
A dose recomendada de Eligard 7,5 mg é mensal.
O medicamento é administrado por via subcutânea formando um depósito sólido e fornece liberação contínua do medicamento por 1 mês.
A dose recomendada de Eligard 22,5 mg é de uma injeção a cada 3 meses (trimestral).
O medicamento é administrado por via subcutânea formando um depósito sólido e fornece liberação contínua do medicamento por 3 meses.
A dose recomendada de Eligard 45 mg é de uma injeção a cada 6 meses (semestral).
O medicamento é administrado por via subcutânea, e fornece liberação contínua do medicamento por 6 meses.
A administração da suspensão injetável de Eligard 7,5 mg fornece 7,5 mg de acetato de leuprorrelina ao paciente.
A suspensão injetável de Eligard 22,5 mg fornece 22,5 mg de acetato de leuprorrelina. A suspensão injetável de Eligard 45 mg fornece 45 mg de acetato de leuprorrelina.
Uso em idosos
Até o momento, não foi comprovada a necessidade de ajuste de dosagem em pacientes idosos.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Eligard?
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Caso você apresente alguma das situações abaixo, seu médico irá tomar as medidas necessárias:
- Se o câncer de próstata tiver se espalhado para as vertebras (metástases vertebrais);
- Se houver obstrução do trato urinário.
Eligard causa na primeira semana de tratamento um aumento temporário nas concentrações de testosterona (hormônio sexual masculino) no sangue.
Durante as primeiras semanas de tratamento você poderá apresentar piora dos sintomas da doença ou poderá apresentar novos sinais e sintomas, incluindo dor óssea, neuropatia (problemas no sistema nervoso), hematúria (presença de sangue na urina) ou obstrução do trato urinário.
Foram observados casos isolados de obstrução da uretra e/ou compressão da medula espinhal, que podem contribuir para paralisia (com ou sem complicações fatais), durante o tratamento paliativo do câncer de próstata avançado com agonistas de LH-RH.
Caso haja desenvolvimento de compressão da medula espinhal ou comprometimento renal, o tratamento padrão dessas complicações deverá ser instituído.
Hiperglicemia e diabetes
Hiperglicemia e um aumento no risco de desenvolvimento de diabetes tem sido reportado em homens recebendo análogos de GNRH.
Monitoração dos níveis glicêmicos e manejo de acordo com a prática clínica deverá ser realizada.
Alterações nos exames laboratoriais
Os resultados de exames das funções da hipófise e dos testículos realizados durante e após o tratamento com Eligard podem ser afetados.
Eventos Cardiovasculares
Foi observado aumento no risco de infarto do miocárdio, morte súbita e acidente vascular cerebral Monitoração cuidadosa de eventos cardiovasculares e manejo de acordo com a prática clínica deverá ser realizada.
Este medicamento pode causar doping.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento de seu médico. Pode ser perigoso para sua saúde.
Reações Adversas
Eligard causa na primeira semana de tratamento um aumento temporário nas concentrações de testosterona (hormônio sexual masculino) no sangue.
Durante as primeiras semanas de tratamento você poderá apresentar piora dos sintomas da doença ou poderá apresentar novos sinais e sintomas,incluindo dor óssea, neuropatia (problemas no sistema nervoso), hematúria (presença de sangue na urina) ou obstrução do trato urinário.
O agravamento dessas condições pode levar a problemas neurológicos como fraqueza, parestesia (sensação de formigamento) dos membros inferiores (pernas) ou piora dos sintomas urinários.
Foram observados casos isolados de obstrução da uretra e/ou compressão da medula espinhal, que podem contribuir para paralisia (com ou sem complicações fatais).
Poderá ocorrer dor no local da injeção reação como queimação, mais raramente prurido (coceira), endurecimento do local e ulceração (lesão na pele).
Alterações na densidade óssea também foram relatadas.
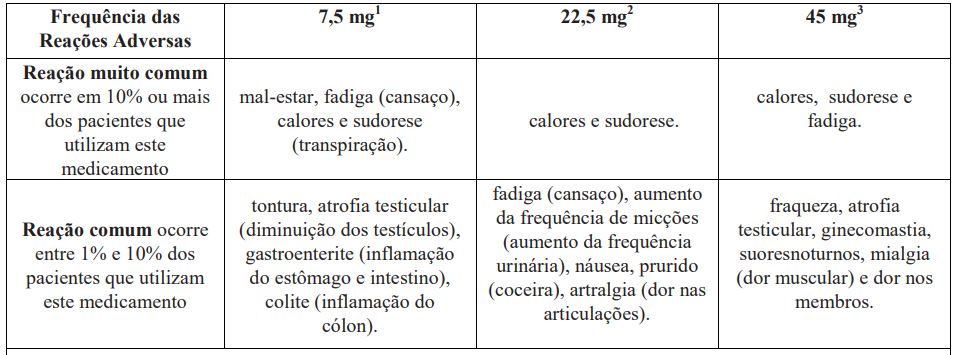
1Outras reações, relatadas em menos de 2% dos pacientes: insônia, síncope (desmaio), flatulência (gases), obstipação (prisão de ventre), redução da contagem de eritrócitos (células vermelhas do sangue), hematócritos (volume total de eritrócitos) e hemoglobina (veículo do oxigênio no sangue), ganho de peso, tremores, dor nas costas, dor articular, distúrbios de olfato e paladar, depressão, vertigem (tontura), alopecia (queda de cabelo), sensibilidade testicular, impotência, redução da libido (diminuição do desejo sexual), ginecomastia (aumento das glândulas mamárias no homem), sensibilidade mamária.
2Outras reações, relatadas em menos de 2% dos pacientes: dispepsia (dificuldade de digestão), tremores, fraqueza, letargia (sonolência / lentidão), dificuldades ao urinar, dor ao urinar, micção curta, espasmos vesicais (contrações na bexiga), hematúria (presença de sangue na urina), retenção urinária (impossibilidade de urinar), maior sensibilidade mamária, atrofia testicular (diminuição dos testículos), dor testicular, ginecomastia (aumento das glândulas mamárias no homem), impotência, suores noturnos, hipertensão (pressão alta) e hipotensão (pressão baixa).
3Outras reações, relatadas em menos de 2% dos pacientes: dispepsia (dificuldade de digestão), sonolência, impotência sexual, desejo de urinar durante o sono, bem como aumento da frequencia urinária durante o sono e perda de desejo sexual.
Eligard 7,5 mg e 22,5 mg
Atenção: este produto é um medicamento que possui nova forma farmacêutica no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico.
Eligard 45 mg
Atenção: este produto é um medicamento que possui nova concentração no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico.
População Especial
Gravidez e amamentação
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Este medicamento causa malformação ao bebê durante a gravidez.
Este medicamento não é indicado para uso em mulheres e não se sabe se ele é excretado no leite materno.
Como muitos medicamentos são excretados no leite materno e devido ao potencial de reações adversas sérias em lactentes (crianças em fase de amamentação) expostos a esta droga, deve-se optar por parar a amamentação se não for possível parar o uso do medicamento.
Essas possibilidades devem ser discutidas com seu médico.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento de seu médico. Pode ser perigoso para sua saúde.
Composição
Eligard 7,5 mg
Eligard 22,5 mg
Eligard 45,0 mg
Superdosagem
Caso ocorra a aplicação de uma superdose recomendam-se medidas gerais de monitorização frequente dos sinais vitais e observação do paciente.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Não foram realizados estudos específicos sobre interação do acetato de leuprorrelina com outras substâncias. Entretanto, considerando que a leuprorrelina é um peptídeo principalmente metabolizado pela peptidase e não pelas enzimas do citocromo P450, conforme observado em estudos específicos, e que a substância é apenas cerca de 46% ligada às proteínas plasmáticas, não são esperadas interações medicamentosas.
Alterações em exames laboratoriais durante o tratamento
A administração de acetato de leuprorrelina em suspensão de depósito em mulheres resulta na supressão do sistema hipofisário-gonadal. A função normal geralmente é recuperada em até 3 meses após a descontinuação do tratamento.
Portanto, os exames de diagnóstico da função hipofisária gonadotrófica e gonadal realizados durante o tratamento até 3 meses após a descontinuação do produto podem não ser conclusivos.
Interação Alimentícia
Não foram realizados estudos, no entanto, não são esperadas reações com com alimentos.
Ação da Substância
Resultados de eficácia
Câncer de próstata
A eficácia de acetato de leuprorrelina foi comprovada em um estudo prospectivo, aberto, para a avaliação da segurança e eficácia do acetato de leuprorrelina, em apresentação de 3,75 mg, mensal, numa população de 205 pacientes com câncer de próstata avançado.
O objetivo principal do estudo era avaliar a eficácia, através da observação dos níveis de testosterona, que deveriam permanecer em níveis de castração (? 50 ng/dL) no período de 45 meses de observação. O nível médio prétratamento de testosterona reduziu de 350 ng/dL para 21 ng/dL após quatro semanas e 20 ng/dL após 45 meses de tratamento.
A eficácia clínica a longo prazo pode ser expressa pela melhor resposta ao tratamento no período: resposta completa – 10,7%; resposta parcial – 49,8%; sem alterações – 34,1%, progressão – 1,5%, sem dados – 3,9%. A mediana de tempo para progressão foi de 12 (15 ± 11) meses.
No estudo I1 foram randomizados 237 pacientes com câncer de próstata avançado ou metastático, tratados com a apresentação mensal do acetato leuprorrelina na concentração de 3,75 mg (grupo 1) ou com a apresentação trimestral do acetato de leuprorrelina 11,25 mg (grupo 2) durante 9 meses de tratamento.
Ambas apresentações produziram efeitos idênticos, com uma queda pronunciada nos níveis séricos de testosterona e gonadotropinas, e redução dos níveis de “PSA” (Antígeno Prostático Específico). Após 9 meses de tratamento, os níveis de “PSA” se normalizaram (? 4 ng/ml) em 65,2% e 66,1% dos pacientes nos grupos 1 e 2 respectivamente.
A resposta clínica, avaliada pelos critérios de remissão da “EORTC” (Organização Européia para Pesquisa e Tratamento do Câncer), foram comparáveis para ambas as apresentações.
| Grupo 1 (3,75 mg mensal) | Grupo 2 (11,25 mg mensal) | |
| N (%) | N (%) | |
| Remissão Completa (RC) | 4 (5,0) | 9 (5,7) |
| Remissão Parcial (RP) | 29 (36,3) | 53 (33,8) |
| Estabilização (E) | 32 (40,0) | 64 (40,8) |
| Progressão | 5 (6,3) | 8 (5,1) |
| Dados perdidos | 10 (12,5) | 23 (14,6) |
| Resposta Global “EORTC” (RC+RP+E) | 65 (81,3) | 126 (80,3) |
No estudo II2 foi avaliada a resposta a longo prazo (43 meses) através do seguimento de uma parte dos pacientes do estudo anterior. Na Alemanha, 62 pacientes fizeram uso de acetato de leuprorrelina na sua apresentação trimestral de 11,25 mg. O estudo de seguimento a longo prazo foi fechado com 37 pacientes deste grupo, pois 25 pacientes faleceram no decorrer do estudo.
Foi conseguido adequada supressão dos níveis séricos de testosterona, o percentual total de supressão em 444 medições realizadas em todos os 62 pacientes durate o período de tratamento foi de 98%.
Ginecologia
Em um estudo farmacocinético/farmacodinâmico de mulheres saudáveis (N=20), foi observado o início da supressão do estradiol nos pacientes entre os dias 04 e a semana 04, após a dose.
Por volta da terceira semana após a injeção, a concentração média do estradiol (8 pg/mL) estava na faixa de menopausa. Ao longo do restante do período de dose, os níveis séricos médios de estradiol variaram da menopausa ao folicular precoce.
O estradiol sérico foi suprimido para ?20 pg/mL em todos os pacientes em quatro semanas de tratamento e permaneceu suprimido (?40 pg/mL) em 80% dos pacientes até o final do intervalo de dose de 12 semanas, quando dois pacientes apresentaram valores entre 40 e 50 pg/mL.
Quatro outros pacientes apresentaram, pelo menos, duas elevações consecutivas dos níveis de estradiol (variação de 43-240 pg/mL) durante o intervalo de dose de 12 semanas, mas não houve indicação de função lútea para qualquer um dos pacientes durante este período.
O acetato de leuprorrelina 11,25 mg, para aplicação a cada 3 meses, induziu amenorreia em 85% (N=17) dos pacientes durante o mês inicial e em 100% deles durante o segundo mês, após a injeção.
Todos os pacientes permaneceram amenorreicos ao longo do restante do intervalo de dose de 12 semanas. Os episódios de sangramento leve e de manchas foram relatados pela maior parte dos pacientes durante o primeiro mês após a injeção e por alguns outros pacientes em épocas posteriores. A menstruação voltou, em média, 12 semanas (variação de 2,9 a 20,4 semanas) após o término do intervalo de dose de 12 semanas.
O acetato de leuprorrelina 11,25 mg, para aplicação a cada 3 meses, produziu efeitos farmacodinâmicos similares em termos de supressão hormonal e menstrual para os pacientes que receberam o acetato de leuprorrelina 3,75 mg durante os estudos clínicos controlados para o manejo da endometriose e da anemia causadas pelos fibromas uterinos.
Endometriose
Em estudos clínicos controlados, o acetato de leuprorrelina 3,75 mg, uma vez ao mês por seis meses, demonstrou que é comparável ao danazol de 800 mg/dia no alívio dos sinais/sintomas clínicos da endometriose (dor pélvica, dismenorreia, dispareunia, sensibilidade pélvica e enrijecimento) e na redução dos implantes endometriais, de acordo com as evidências de laparoscopia.
A significância clínica da diminuição de lesões endometriais atualmente não é conhecida, além disso, o estadiamento laparoscópico não necessariamente se correlaciona à severidade dos sintomas.
Acetato de leuprorrelina 3,75 mg, mensalmente, induziu a amenorreia em 74% e 98% dos pacientes após o primeiro e o segundo mês de tratamento, respectivamente. A maior parte dos pacientes relatou episódios de sangramento leve ou de manchas. No primeiro, no segundo e no terceiro mês pós-tratamento, os ciclos menstruais normais retornaram em 7%, 71% e 95% dos pacientes, respectivamente, exceto pelas pacientes que engravidaram.
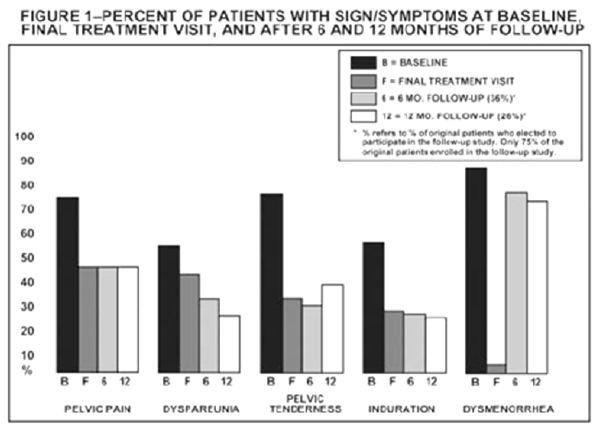
A Figura a seguir ilustra a porcentagem de pacientes com sintomas no baseline, na visita final de tratamento e o alívio sustentado aos 6 e aos 12 meses após a descontinuação do tratamento para os diversos sintomas avaliados durante os dois estudos clínicos controlados, incluindo todos os pacientes no final do tratamento e os que se elegeram a participar do período de acompanhamento.
Pode-se levar a uma tendência leve nos resultados do período de acompanhamento, pois 75% dos pacientes originais iniciaram o estudo de acompanhamento, 36% foram avaliados aos 6 meses e 26% aos 12 meses.
Leiomioma Uterino (Fibroma uterino)
Em estudos clínicos controlados, a administração de acetato de leuprorrelina 3,75 mg por três ou seis meses demonstrou reduzir o volume uterino e do fibroma, portanto, permitindo alívio dos sintomas clínicos (inchaço abdominal, dor pélvica e pressão).
O sangramento vaginal excessivo (menorragia e menometrorragia) diminuiu resultando em melhora dos parâmetros hematológicos. Em três estudos clínicos, o registro dos pacientes não se baseou na condição hematológica. O volume do útero diminuiu em 41% e o volume do mioma diminuiu em 37% na visita final, de acordo com as evidências do ultrassom ou imagens por ressonância magnética (MRI).
Além disso, esses pacientes apresentaram uma diminuição dos sintomas, incluindo o sangramento vaginal excessivo e o desconforto pélvico. O benefício ocorreu por volta dos três meses de terapia, mas foi observado ganho adicional com mais três meses de terapia com acetato de leuprorrelina 3,75 mg.
Noventa e cinco porcento (95%) desses pacientes tornaram-se amenorreicos, com 61%, 25%, e 4% com amenorreia no primeiro, segundo e terceiro mês de tratamento, respectivamente. O acompanhamento pós-tratamento foi realizado com uma porcentagem pequena de pacientes que receberam acetato de leuprorrelina 3,75 mg (N =46) dentre os 77% que demonstraram um aumento ? 25% no volume do útero durante a terapia.
A menstruação, de um modo geral, retornou em duas semanas de supressão de terapia. O tempo médio para o retorno ao tamanho do útero pré-tratamento foi de 8,3 meses. O novo crescimento não pareceu estar relacionado com o volume de útero no pré-tratamento.
Em outro estudo clinico controlado, o registro dos pacientes baseou-se no hematócrito ? 30% e/ou hemoglobina ? 10,2 g/dL. A administração de acetato de leuprorrelina 3,75 mg, concomitante com o ferro, produziu um aumento de ? 6% no hematócrito e de ? 2 g/dL em hemoglobina em 77% dos pacientes aos três meses de terapia. A alteração média no hematócrito foi de 10,1% e a alteração média na hemoglobina foi de 4,2 g/dL.
Foi julgado que a resposta clínica ocorreria em um hematócrito de ? 36% e hemoglobina de ? 12 g/dL, permitindo, portanto, a doação autóloga de sangue antes da cirurgia. Aos três meses, 75% dos pacientes cumpriram esse critério.
Aos três meses, 80% dos pacientes experimentaram alívio ou da menorragia ou da menometrorragia. Assim como nos estudos anteriores, foram notados episódios de manchas e de sangramento do tipo menstrual em alguns dos pacientes.
Nesse mesmo estudo, foi observada uma diminuição de ? 25% no volumes do útero e do mioma em 60% e 54% dos pacientes, respectivamente. Foi observado que acetato de leuprorrelina 3,75 mg aliviou os sintomas de inchaço, de dor pélvica e de pressão.
Não há evidências de que as taxas de gravidez aumentem ou que sejam afetadas adversamente pelo uso de acetato de leuprorrelina 3,75 mg.
Câncer de mama
A eficácia do acetato de leuprorrelina no tratamento do câncer de mama avançado ficou comprovada no estudo prospectivo, em 76 pacientes na perimenopausa. Foram feitas doses mensais de acetato de leuprorrelina 3,75 mg, até se observar a progressão da doença.
Foram feitas análises de resposta aos 3 e 6 meses de tratamento. Resposta objetiva foi alcançada em 39 (51,3%) das pacientes após 3 meses em 23 (30,3%) dos pacientes após 6 meses, e
foram definidas como remissão completa ou parcial ou estabilização da doença de acordo com os critérios da OMS utilizados na época (OMS 1979).
A eficácia do acetato de leuprorrelina de 11,25 mg no tratamento do câncer de mama avançado ficou comprovada no estudo prospectivo, de fase III, randomizado, aberto, multicêntrico, que teve como comparador pacientes que eram submetidas a quimioterapia padrão com ciclofosfamida, metotrexato e fluouracil (CMF).
No estudo TABLE3, 599 pacientes em pré-menopausa com diagnóstico de câncer de mama, positivos para receptor de estrogênio, confirmados histologicamente, nos estágios II ou IIIA, foram elegíveis para o estudo e randomizados, dentro de 6 semanas após o tratamento cirúrgico, em dois grupos de tratamento.
No grupo 1 foram tratadas com o acetato de leuprorrelina 11,25 mg, 299 pacientes, e no grupo 2 foram tratadas com o esquema quimioterápico padrão CMF, 300 pacientes. O seguimento médio foi de 5,8 anos, a sobrevida livre de recidiva foi similar em ambos os grupos de tratamento (P =0.15). Uma análise exploratória da sobrevida global foi favorável ao grupo de tratamento com acetato de leuprorrelina de 11,25 mg, com uma taxa de sobrevida em 5 anos de 81% para o grupo 1 e de 71,9% para o grupo 2 (P =0.005).
Também houve uma tendência maior de mortalidade relacionada ao câncer de mama no grupo 2.
Os resultados desse estudo demonstraram que o tratamento com acetato de leuprorrelina de 11,25 mg, de uso trimestral é tão efetivo, e não é inferior ao tratamento quimioterápico padrão com CMF, nesta população de pacientes com diagnóstico de câncer de mama.
Seus dados de eficácia podem também ser interpretados como dados que dão sustentação para a preparação mensal de 3,75 mg.
Puberdade Precoce
Nas crianças com puberdade precoce central (CPP), as gonadotrofinas estimuladas e basais são reduzidas para os níveis pré-puberdade. A testosterona e o estradiol são reduzidos aos níveis pré-puberdade nos meninos e nas meninas, respectivamente.
A redução das gonadotrofinas permitirá o crescimento e o desenvolvimento físico e psicológico normais.
A maturação natural ocorre quando as gonadotrofinas voltam aos níveis pré-puberdade após a descontinuação de acetato de leuprorrelina.
Um estudo foi realizado com um grupo de 55 indivíduos com puberdade precoce central (49 do sexo feminino e 6 do sexo masculino, nunca tratados anteriormente com agonistas de GnRH), tratados mensalmente com acetato de leuprorrelina até atingir a idade apropriada para a puberdade. Após a interrupção do tratamento, foi feito o acompanhamento de 40 indivíduos deste grupo.
Foram notados os seguintes efeitos fisiológicos com a administração crônica do acetato de leuprorrelina nessa população de pacientes:
Dados do Período de Tratamento
Durante o período de tratamento, a administração mensal de acetato de leuprorrelina suprimiu os níveis de gonadotropinas e esteróides sexuais a níveis pré-puberais. A supressão das concentrações de pico estimuladas por LH para valores < 1,75 mIU/mL foi atingida por 96% dos indivíduos no mês 1.
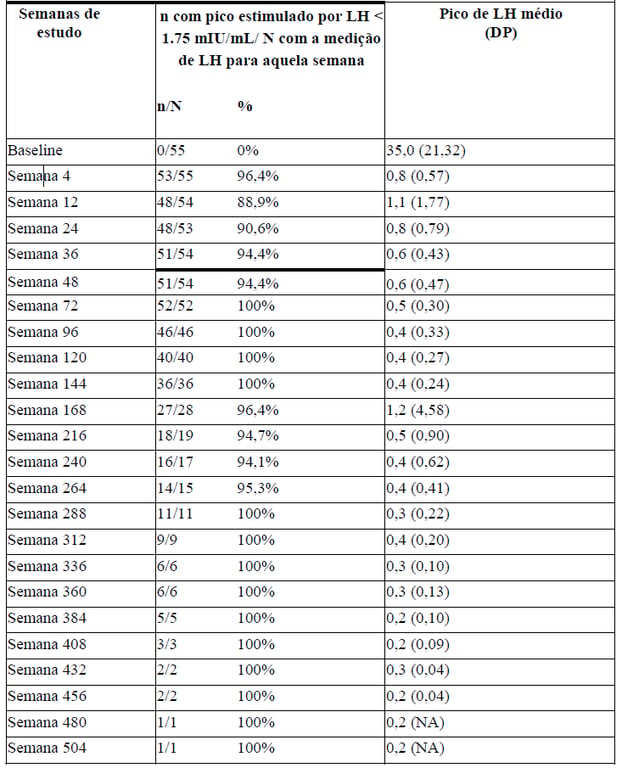
O número e a porcentagem de indivíduos com supressão do pico estimulado por LH < 1,75 mIU/mL e o pico médio + desvio padrão do pico estimulado por LH ao longo do tempo é apresentado na Tabela 1.
A idade média + DP no início do tratamento foi 7 + 2 anos e a duração do tratamento foi 4 + 2 anos. Seis meses após o fim do período de tratamento, o pico médio estimulado por LH foi de 20,6 + DP 13,7 mIU/mL (n=30).
Tabela 1: Número e porcentagem de pacientes com pico estimulado por LH < 1,75 mIU/mL e Pico de LH médio (DP) em cada visita clínica
A supressão (definida como regressão ou não mudança) dos sinais clínicos/físicos da puberdade foi atingida pela maioria dos pacientes. Em mulheres, a supressão do desenvolvimento das mamas variou de 66,7 a 90,6% dos indivíduos durante os primeiros 5 anos de tratamento.
A média do estradiol estimulado foi de 15,1pg/mL nas condições basais, diminuindo para o menor nível de detecção (5,0 pg/mL) na semana 4 e foi mantido durante os primeiros 5 anos de tratamento. Em homens, a supressão do desenvolvimento da genitália variou de 60% a 100% dos indivíduos durante os primeiros 5 anos de tratamento.
A média de testosterona estimulada foi 347,7 ng/dL nas condições basais e foi mantida a níveis não superiores a 25,3 ng/dL durante os 5 primeiros anos de tratamento.
Um “efeito rebote” da hemorragia transitória ou sangramento vaginal durante as 4 primeiras semanas de tratamento foi observado em 19,4% (7/36) meninas que não atingiram a menarca nas condições basais. Após as primeiras 4 semanas e durante o período de tratamento restante, nenhum indivíduo relatou sangramento semelhante à menstruação, e apenas um raro sangramento vaginal foi observado.
Em muitos indivíduos, houve diminuição da taxa de crescimento durante o tratamento, assim como a razão idade óssea: idade cronológica. Após 5 anos, a taxa de crescimento média variou entre 3,4 a 5,6 cm/ano. A razão média da idade óssea pela idade cronológica diminuiu de 1,5 nas condições basais para 1,1 no final do tratamento.
A pontuação de desvio padrão da estatura média mudou de 1,6 nas condições basais para 0,7 no fim da fase de tratamento.
Dados do período do acompanhamento
Para a avaliação da função reprodutiva (em mulheres) e da estatura definitiva, 35 mulheres e 5 homens participaram de um período de acompanhamento pós-tratamento. Aos 6 meses pós-tratamento, a maioria dos indivíduos apresentou reversão dos níveis puberais de LH (87,9%) e os sinais clínicos de retomada da progressão puberal foram evidentes com o aumento no desenvolvimento das mamas em garotas (66,7%) e aumento do desenvolvimento da genitália em garotos (80%).
Dos 40 pacientes avaliados no acompanhamento, 33 foram observados até que eles atingissem estatura adulta definitiva ou quase definitiva. Estes pacientes apresentaram um aumento médio da estatura adulta definitiva quando comparado à estatura adulta prevista nas condições basais.
A pontuação de desvio padrão da estatura adulta definitiva média foi -0,2. Após a interrupção do tratamento, foram relatadas menstruações regulares para todos os indivíduos do sexo feminino que atingiram 12 anos durante o acompanhamento; o tempo médio para a menstruação foi de aproximadamente 1,5 anos; a idade média no início da menstruação foi 12,9 anos.
Os dados para avaliar a função reprodutiva foram obtidos em um exame pós-estudo de 20 garotas que atingiram a maioridade (18-26 anos); foram relatados ciclos menstruais normais em 80% das mulheres; 12 casos de gravidez foram relatados de 7 dos 20 indivíduos, incluindo múltiplos casos de gravidez para 4 indivíduos.
Em um estudo clínico randomizado e aberto realizado com acetato de leuprorrelina 11,25 mg em formulação para uso trimestral, 42 pacientes, com idade entre 1 e 11 anos, receberam o medicamento. O grupo estudado teve um número igual de pacientes virgens de tratamento que tinham níveis de LH puberais e pacientes que estavam em tratamento prévio com agonistas do GnRHa mensais que tinham níveis de LH pré-puberais ao início do estudo.
O percentual de pacientes com supressão de níveis de pico de LH estimulado para < 4 mUI/mL, como determinado por meio de avaliações nos meses 2, 3 e 6 foi de 78,6%.
Tabela 2: Supressão de níveis de pico de LH estimulado entre o mês 2 e o mês 6

A média de pico de LH estimulada para todas as visitas é mostrada no gráfico abaixo por dose e subgrupo (virgens de tratamento e previamente tratados)
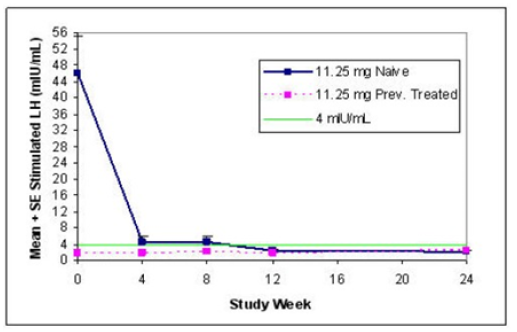
O porcentual de 93% (39 de 42) dos pacientes tiveram os níveis de esteróides sexuais (estradiol e testosterona) suprimidos à níveis pré-puberais em todas as visitas.
Supressão clínica da puberdade em pacientes do sexo feminino foi observada em 29 de 32 (90,6%) pacientes no mês 6.
Supressão clínica da puberdade em pacientes do sexo masculino foi observada em 1 de 2 pacientes (50,0%) no mês 6. Em pacientes com informações completas relacionadas à idade óssea, 29 de 33 sujeitos (87,9%) tiveram uma diminuição na razão entre idade óssea e idade cronológica no mês 6 quado comparado à triagem.
Características Farmacológicas
Descrição
O acetato de leuprorrelina, substância ativa dest medicamento (acetato de leuprorrelina), é um nonapeptídeo sintético análogo do hormônio liberador da gonadotrofina natural (GnRH ou LH-RH). Possui maior potência que o hormônio natural, atua como um inibidor da produção de gonadotrofina e é quimicamente distinto dos esteroides. Seu nome químico é acetato de 5-oxo-L-prolil-L-histidil-L-triptofanil-L-seril-L-tirosil-D-leucil-L-leucil-L-arginil-L-Netil-L-prolinamida (sal). O acetato de leuprorrelina neste medicamento (acetato de leuprorrelina) é apresentado como microesferas liofilizadas estéreis que, quando misturadas com o diluente, tornam-se uma suspensão para uso intramuscular.
Farmacodinâmica
O acetato de leuprorrelina - um agonista GnRH - age como um potente inibidor da secreção de gonadotropina, quando administrado continuamente e em doses terapêuticas. Os estudos em animais e em humanos indicam que, seguindo-se a uma estimulação inicial de gonadotropinas, a administração crônica de acetato de leuprorrelina resulta em suspensão da esteroidogênese ovariana e testicular. Esse efeito é reversível com a descontinuação da terapêutica.
A administração de acetato de leuprorrelina resultou na inibição de crescimento de tumores hormônio-dependentes (tumores prostáticos em ratos machos das espécies Noble e Dunning e tumores mamários DMBA-induzidos em ratas), assim como em atrofia de órgãos reprodutivos.
Em humanos, a administração de acetato de leuprorrelina resulta num aumento inicial dos níveis circulantes do hormônio luteinizante (LH) e do hormônio folículoestimulante (FSH), conduzindo a um transitório aumento dos níveis dos esteroides gonadais (testosterona e diidrotestosterona em homens; e estrona e estradiol em mulheres na pré-menopausa).
Contudo, a administração contínua do acetato de leuprorrelina, nas doses recomendadas, resulta em diminuição dos níveis de LH, FSH e esteroides sexuais.
Em homens, a testosterona é reduzida aos níveis de castração. Em mulheres na pré-menopausa, os estrógenos são reduzidos aos níveis pós-menopausa. A redução dos níveis desses hormônios ocorre dentro de um mês após o início do tratamento.
Farmacocinética
O acetato de leuprorrelina não é ativo quando administrado por via oral. A biodisponibilidade quando administrado por via subcutânea é comparável à da administração intramuscular. A biodisponibilidade absoluta de acetato de leuprorrelina 7,5 mg é estimada em 90%.
Absorção
Os níveis séricos médios obtidos ao final de 1 mês após administração única de acetato de leuprorrelina, em pacientes com neoplasia prostática, nas doses de 3,75 mg ou 7,5 mg, por via subcutânea ou intramuscular, foram respectivamente de 0,7 ng/mL e 1,0 ng/mL.
Não houve indícios de acúmulo do fármaco no organismo. Observou-se, em um estudo com pacientes masculinos orquiectomizados, concentrações plasmáticas de acetato de leuprorrelina por período superior a 1 mês após a administração intramuscular de acetato de leuprorrelina 7,5 mg.
Similarmente, em outro estudo envolvendo pacientes com carcinoma prostático em estágio D2, detectaram-se níveis sistêmicos de acetato de leuprorrelina 4 semanas após a administração de uma única dose de acetato de leuprorrelina 7,5 mg.
Distribuição
O volume de distribuição da leuprorrelina, no estado de equilíbrio, após administração intravenosa, em bolus, em voluntários sadios do sexo masculino foi de 27 litros. A ligação às proteínas plasmáticas humanas in vitro variou de 43% a 49%.
Metabolismo
Em voluntários sadios do sexo masculino, uma injeção de 1 mg de leuprorrelina por via intravenosa em bolus, revelou que a depuração sistêmica média foi de 7,6 L/h, com meia vida de eliminação final de aproximadamente três horas, com base em um modelo de dois compartimentos.
Estudos em animais mostraram que a leuprorrelina marcada com C14 foi metabolizada em peptídeos menores inativos, um pentapeptídeo (Metabólito I), tripeptídeos (Metabólitos II e III) e um dipeptídeo (Metabólito IV). Esses fragmentos podem ser metabolizados posteriormente.
As concentrações plasmáticas do principal metabólito (M-I), avaliadas em cinco pacientes com câncer de próstata que receberam acetato de leuprorrelina em suspensão de depósito, atingiram a concentração máxima em duas a seis horas depois da administração e foram aproximadamente 6% da concentração de pico da substância-mãe. Uma semana depois da administração, as concentrações plasmáticas médias de M-I foram aproximadamente 20% das concentrações médias da leuprorrelina.
Eliminação
Após a administração de 3,75 mg do acetato de leuprorrelina em suspensão de depósito a três pacientes, menos de 5% da dose administrada foi recuperada sob a forma de substância-mãe e metabólito M-I na urina em 27 dias.
Populações especiais
A farmacocinética do acetato de leuprorrelina não foi determinada em pacientes com insuficiência hepática ou insuficiência renal.
Cuidados de Armazenamento
Eligard deve ser conservado sob refrigeração (2º a 8ºC).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparo, usar em até 30 minutos.
Depois deste período, a suspensão injetável não utilizada deverá ser descartada.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características organolépticas
Após preparo a suspensão apresentará uma coloração amarela clara a amarela.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Dizeres Legais
MS - 1.2214.0093
Resp Téc.:
Marcia da Costa Pereira
CRF-SP no 32.700.
Fabricado por:
Aptalis Pharmatech, Inc.
Vandalia Ohio 45377.
Estados Unidos.
Importado por:
Zodiac produtos farmacêuticos S/A.
Rodovia Vereador Abel Fabrício Dias,
3400 Pindamonhangaba - SP.
C.N.P.J. 55.980.684/0001-27
Indústria Brasileira.
Venda sob prescrição médica.
informações complementares
| Fabricante |
| ZODIAC |
| Princípio ativo |
| Acetato De Leuprorrelina |
| Categoria do medicamento |
| Medicamentos de A-Z |