Comparamos o preço de Hetori - 90 Mg 7 Comprimidos Revestidos, veja o menor preço

R$ 83,13
ISimilar Intercambiável
1
ofertasMelhores preços a partir de R$ 83,13 até R$ 83,13
Menor preço
vendido por promofarma
R$ 83,13
Para que serve
Hetori é indicado para:
- Tratamento da osteoartrite;
- Tratamento da artrite reumatoide;
- Tratamento de espondilite anquilosante (inflamação da coluna e de grandes articulações);
- Alívio da dor;
- Tratamento de dor aguda após cirurgia dentária;
- Tratamento de dor aguda após cirurgia ginecológica abdominal.
Como o Hetori funciona?
Hetori faz parte de um grupo de medicamentos denominados coxibes, usados para diminuir a dor e a inflamação. Hetori é um inibidor seletivo da COX-2. Hetori não é um narcótico.
Nosso organismo produz dois tipos de enzimas semelhantes, denominadas COX-1 e COX-2. Uma das funções da COX-1 está relacionada com a proteção do estômago, enquanto a COX-2 participa nos processos inflamatórios e dolorosos de tecidos e das articulações.
- Hetori bloqueia a COX-2 e, desse modo, reduz a dor e a inflamação.
- Hetori não bloqueia a COX-1, substância envolvida na proteção contra úlceras no estômago.
- Outros anti-inflamatórios (anti-inflamatórios não esteroides – AINEs) bloqueiam tanto a COX-1 como a COX-2.
- Hetori alivia a dor e a inflamação com menos risco de úlceras no estômago em comparação com os AINEs.
Informações sobre as doenças
As doenças das articulações (também conhecidas como “juntas”) são popularmente chamadas de reumatismo e podem se apresentar de várias formas (por exemplo, osteoartrite, artrite reumatoide, gota, etc.).
O que é osteoartrite?
A osteoartrite é uma doença das articulações. É o resultado da destruição gradual da cartilagem que envolve as extremidades dos ossos causando dor, inflamação, aumento da sensibilidade, rigidez e perda da função da articulação.
O que é artrite reumatoide?
A artrite reumatoide é uma doença crônica que causa dor, rigidez, inchaço e perda da função das articulações e inflamação de outros órgãos do corpo.
O que é espondilite anquilosante?
A espondilite anquilosante é uma doença inflamatória da coluna e de grandes articulações (por exemplo, quadril, joelho e ombro).
O que posso fazer para ajudar a controlar essas doenças, além de tomar remédios?
Converse com seu médico sobre:
- Exercícios físicos;
- Controle de peso;
- Fisioterapia;
- Uso de instrumentos de apoio.
O que é cirurgia ginecológica abdominal?
Qualquer cirurgia abdominal na área do útero (ventre) e/ou de outros órgãos femininos.
Contraindicação
Você não deve tomar Hetori se:
- For alérgico a qualquer um de seus componentes;
- Tiver histórico de insuficiência cardíaca, ataque cardíaco, cirurgia de revascularização (por exemplo, ponte de safena), dor no peito (angina), estreitamento ou bloqueio de artérias das extremidades do corpo (doença arterial periférica), derrame ou derrame transitório (ataque isquêmico transitório - AIT).
Como usar
Hetori deve ser tomado uma vez ao dia, com ou sem alimentos.
Seu médico decidirá a dose e por quanto tempo você deverá tomar Hetori, de acordo com os critérios abaixo.
Para o tratamento da osteoartrite:
A dose recomendada é de 60 mg uma vez ao dia.
Para o tratamento da artrite reumatoide:
A dose recomendada é de 90 mg uma vez ao dia.
Para o tratamento da espondilite anquilosante:
A dose recomendada é de 90 mg uma vez ao dia.
Para alívio da dor crônica:
A dose recomendada é de 60 mg uma vez ao dia.
Condições de dor aguda:
A dose recomendada é de 90 mg uma vez ao dia. Hetori deve ser utilizado apenas durante o período agudo, limitado a 8 dias no máximo.
Para alívio da dor após cirurgia dentária:
A dose recomendada é de 90 mg uma vez ao dia, limitada ao máximo de 3 dias de tratamento.
Para alívio de dor após cirurgia ginecológica abdominal:
A dose recomendada é de 90 mg uma vez ao dia, limitada ao máximo de 5 dias de tratamento. A primeira dose deve ser tomada logo antes da cirurgia.
Doses maiores que as recomendadas para cada situação citada acima não devem ser utilizadas. Caso você tenha uma doença leve do fígado, deve tomar no máximo 60 mg ao dia; se tiver doença moderada do fígado, deve tomar no máximo 60 mg em dias alternados.
Não dê seus comprimidos de Hetori para outra pessoa. Eles foram prescritos por seu médico somente para você.
O que devo fazer quando eu me esquecer de usar o Hetori?
Tente tomar Hetori de acordo com a receita do seu médico. No entanto, caso você se esqueça de tomar uma dose, não tome uma dose extra. Apenas retome o tratamento no dia seguinte.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
- Em estudos clínicos, o risco de desenvolvimento de úlceras com etoricoxibe foi menor do que com AINEs. Nesses estudos algumas pessoas desenvolveram úlceras ao tomar etoricoxibe ou placebo, no entanto a incidência foi mais alta naqueles que tomaram etoricoxibe.
- Caso você sinta falta de ar, dor no peito ou inchaço no tornozelo ou algum desses sintomas piorar interrompa o tratamento com Hetori e consulte um médico assim que possível.
- Se você tiver doença no rim, no fígado ou no coração, seu médico irá acompanhá-lo apropriadamente.
- Se você desenvolver algum sintoma indicativo de reação alérgica grave, como dificuldade para respirar ou reação grave na pele, procure um médico imediatamente.
- Seu médico poderá discutir seu tratamento com você de tempos em tempos. É importante que você tome a dose mais baixa suficiente para controlar sua dor e que não tome Hetori por mais tempo que o necessário. Isso se deve ao fato de que o risco de ataques cardíacos e derrames pode aumentar após o tratamento prolongado, especialmente com altas doses.
- Hetori pode aumentar a pressão arterial em algumas pessoas, especialmente em altas doses, o que pode aumentar o risco de ataques cardíacos e derrames. Seu médico irá verificar sua pressão regularmente para se certificar de que é seguro continuar o tratamento.
Informe ao médico qualquer problema de saúde ou alergia que você apresente ou já tenha apresentado, incluindo:
- Doenças do coração, tais como angina, ataque cardíaco ou bloqueio de artéria no coração;
- Dstreitamento ou bloqueio das artérias das extremidades;
- Doenças dos rins;
- Doenças do fígado;
- Desidratação, por exemplo, uma crise prolongada de vômitos ou diarreia;
- Histórico de sangramento no estômago ou úlcera;
- Insuficiência cardíaca;
- Inchaço devido a retenção de líquidos;
- Pressão alta;
- Reação alérgica ao ácido acetilsalicílico ou a outros anti-inflamatórios (geralmente conhecidos como AINEs - anti-inflamatórios não esteroides);
- Histórico de derrame ou derrame transitório;
- Condições que aumentem o risco de doença arterial coronariana ou aterosclerose, como pressão alta, diabetes, colesterol alto ou tabagismo,
- Caso esteja recebendo tratamento para alguma infecção. Hetori pode mascarar ou esconder a febre, que é um sinal de infecção.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Qualquer medicamento pode apresentar efeitos inesperados ou indesejáveis, denominados efeitos adversos, e Hetori também pode apresentá-los.
Se você desenvolver qualquer um dos sinais abaixo, deve parar de tomar Hetori e falar com seu médico imediatamente:
- Falta de ar, dores no peito ou inchaço no tornozelo ou agravamento desses sintomas;
- Amarelamento da pele e dos olhos (icterícia) - estes são sinais de problemas no fígado;
- Dor abdominal grave ou persistente ou se as fezes se tornarem negras;
- Reações alérgicas - que podem incluir problemas de pele, tais como úlceras ou vesículas ou inchaço da face, lábios, língua ou garganta que podem causar dificuldade para respirar.
Os seguintes efeitos adversos podem ocorrer durante o tratamento com Hetori:
Muito Comum (ocorre em pelo menos 10% dos pacientes que utilizam este medicamento):
Alveolite (inflamação e dor após extração dentária).
Comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento):
Fraqueza e fadiga, tontura, dor de cabeça, doença semelhante à gripe, diarreia, flatulência, náuseas, má digestão (dispepsia), dor ou desconforto no estômago, azia, alterações nos exames de sangue relacionados ao seu fígado, inchaço das pernas e/ou pés devido à retenção de líquidos (edema), aumento da pressão arterial, palpitações e hematomas.
Incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento):
Distensão abdominal do estômago ou intestino, dor no peito, insuficiência cardíaca; sensação de aperto, pressão ou peso no peito (angina), ataque cardíaco, derrame, derrame transitório, batimento anormal do coração (fibrilação atrial), infecção do trato respiratório superior, altos níveis de potássio no sangue, alterações no sangue ou urina relacionadas aos rins, alterações nos hábitos intestinais, incluindo prisão de ventre, boca seca, aftas, alteração do paladar, gastroenterite, gastrite, úlcera gástrica, enjoo (vômitos), síndrome do intestino irritável, inflamação do esôfago, visão turva, irritação e vermelhidão nos olhos, sangramento nasal, zumbido nos ouvidos, vertigens, aumento ou diminuição do apetite, ganho de peso, câimbras/espasmos, dor muscular/rigidez, incapacidade de dormir, insônia, redução da sensibilidade ou formigamento nas extremidades, ansiedade, depressão, redução da agilidade mental, falta de ar, tosse, inchaço da face, rubor, erupção cutânea ou coceira, infecção do trato urinário, diminuição de plaquetas, diminuição do número de glóbulos vermelhos, diminuição do número de glóbulos brancos.
Raro (ocorre entre 0,01% e 0,1% dos pacientes que utilizam este medicamento):
Baixos níveis sanguíneos de sódio, vermelhidão na pele.
Muito raro (ocorre em menos de 0,01% dos pacientes que utilizam este medicamento):
Reações alérgicas (que podem ser suficientemente graves para exigir assistência médica imediata) incluindo urticária, inchaço da face, lábios, língua e/ou garganta que podem causar dificuldade para respirar ou engolir, broncoespasmo (chiado ou falta de ar), reações graves na pele, inflamação da parede do estômago ou úlceras que possam se agravar e provocar sangramentos, problemas no fígado, problemas graves nos rins, aumento grave da pressão sanguínea, confusão, ver, sentir ou ouvir coisas que não existem (alucinações).
Desconhecido (a frequência não pode ser estimada a partir dos dados disponíveis)
Amarelamento da pele e dos olhos (icterícia), inflamação do pâncreas, batimento cardíaco acelerado, ritmo cardíaco irregular (arritmia), agitação, insuficiência hepática.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento.
População Especial
Gravidez e Amamentação:
Informe ao seu médico se estiver grávida ou se pretender engravidar; informe ao seu médico se estiver amamentando ou se pretender amamentar.
Hetori não deve ser utilizado por mulheres com gravidez avançada, porque pode causar danos ao feto.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Uso pediátrico:
Hetori não foi adequadamente estudado em crianças, portanto Hetori não é recomendado a crianças.
Idosos:
Hetori age da mesma forma nos pacientes idosos e nos adultos mais jovens. As experiências adversas podem ocorrer com maior frequência nos pacientes idosos em comparação com os mais jovens. Se tiver mais de 65 anos de idade, seu médico irá avaliá-lo e acompanhá-lo apropriadamente. Não é necessário ajuste da dose para os pacientes idosos.
Dirigir ou Operar Máquinas:
Não existem informações sugestivas de que o uso de Hetori possa afetar sua capacidade de dirigir veículos ou operar máquinas.
Composição
Cada comprimido revestido contém:
Toricoxibe: 60 mg ou 90 mg.
Excipientes: fosfato de cálcio dibásico anidro, cera de carnaúba, croscarmelose sódica, hipromelose, lactose monoidratada, estearato de magnésio, celulose microcristalina, dióxido de titânio e triacetina. Hetori 60 mg também contém óxido férrico (amarelo) e FD&C azul número 2 (laca índigo carmim).
Superdosagem
Se você tomar mais do que a dose prescrita, procure um médico imediatamente.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Varfarina:
Em indivíduos estabilizados sob tratamento crônico com a varfarina, a administração de Etoricoxibe (substância ativa) 120 mg uma vez ao dia foi associada a aproximadamente 13% de aumento no tempo de protrombina (International Normalized Ratio – INR). Em pacientes que estejam recebendo varfarina ou agentes semelhantes, deve-se proceder a monitorização-padrão dos valores de INR ao se iniciar ou alterar o tratamento com Etoricoxibe (substância ativa), principalmente nos primeiros dias de tratamento.
Rifampicina:
A coadministração de Etoricoxibe (substância ativa) e rifampicina, um potente indutor do metabolismo hepático, reduziu em 65% a área sob a curva da concentração plasmática (AUC) do Etoricoxibe (substância ativa). Essa interação deve ser considerada quando Etoricoxibe (substância ativa) for administrado com rifampicina.
Metotrexato:
Dois estudos pesquisaram os efeitos de Etoricoxibe (substância ativa) nas doses de 60 mg, 90 mg e 120 mg administradas uma vez ao dia durante sete dias em pacientes com artrite reumatoide que recebiam doses de 7,5 mg a 20 mg de metotrexato uma vez por semana. Em um estudo, as doses de Etoricoxibe (substância ativa) 60 mg e 90 mg não exerceram efeito na concentração plasmática do metotrexato (conforme avaliado pela AUC) ou na depuração renal, o que também ocorreu com a dose de Etoricoxibe (substância ativa) 120 mg em um estudo. Em outro estudo, Etoricoxibe (substância ativa) na dose de 120 mg aumentou a concentração plasmática do metotrexato em 28% (conforme avaliado pela AUC) e reduziu a depuração renal do metotrexato em 13%. A monitorização da toxicidade relacionada ao metotrexato deve ser considerada quando este for administrado concomitantemente a doses maiores de 90 mg ao dia de Etoricoxibe (substância ativa).
Diuréticos, Inibidores da Enzima Conversora de Angiotensina (ECA) e Bloqueadores do Receptor de Angiotensina II (BRAs):
Relatos sugerem que os AINEs, incluindo os inibidores seletivos da COX-2, podem diminuir o efeito anti-hipertensivo dos diuréticos, dos inibidores da ECA e dos BRAs. Essa interação deve ser considerada quando Etoricoxibe (substância ativa) for administrado concomitantemente com esses produtos.
Em alguns pacientes com função renal comprometida (por exemplo, pacientes idosos ou pacientes com hipovolemia, incluindo pacientes sob tratamento diurético) que estejam sendo tratados com AINEs, incluindo inibidores seletivos da COX-2, a administração concomitante de inibidores da ECA ou de BRAs pode resultar em deterioração adicional da função renal, inclusive possível insuficiência renal aguda. Esses efeitos são em geral reversíveis. Portanto a combinação deve ser administrada com cuidado, particularmente em idosos.
Lítio:
Relatos sugerem que os AINEs não seletivos e os inibidores seletivos da COX-2 podem aumentar os níveis plasmáticos de lítio. Essa interação deve ser considerada quando Etoricoxibe (substância ativa) for administrado concomitantemente com lítio.
Ácido Acetilsalicílico:
Etoricoxibe (substância ativa) pode ser administrado concomitantemente com ácido acetilsalicílico em baixas doses para profilaxia cardiovascular. Em estado de equilíbrio, a dose de 120 mg de Etoricoxibe (substância ativa) uma vez ao dia não exerceu efeito na atividade antiplaquetária do ácido acetilsalicílico em baixas doses (81 mg uma vez ao dia).
Entretanto, a administração concomitante de Etoricoxibe (substância ativa) com baixas doses de ácido acetilsalicílico aumenta a incidência de ulceras ou de outras complicações do trato gastrintestinal em comparação com o uso de Etoricoxibe (substância ativa) isoladamente.
Anticoncepcionais Orais:
A administração concomitante de Etoricoxibe (substância ativa) 60 mg e de um anticoncepcional oral com 35 mcg de etinilestradiol e 0,5 a 1 mg de noretindrona durante 21 dias aumentou a AUC0-24h no estado de equilíbrio do etinilestradiol em 37%; a administração concomitante, ou com intervalo de 12 horas, de Etoricoxibe (substância ativa) 120 mg com o mesmo anticoncepcional oral aumentou a AUC0-24h no estado de equilíbrio do etinilestradiol em 50% a 60%.
Esse aumento da concentração do etinilestradiol deve ser considerado ao se escolher um anticoncepcional oral para ser utilizado com Etoricoxibe (substância ativa). Um aumento na exposição ao etinilestradiol pode aumentar a incidência de eventos adversos associados aos anticoncepcionais orais (por exemplo, eventos tromboembólicos venosos em mulheres com risco).
Terapia de Reposição Hormonal:
A administração de Etoricoxibe (substância ativa) 120 mg com estrogênios conjugados para terapia de reposição hormonal (0,625 mg de estrogênios conjugados), durante 28 dias, aumentou a média da AUC0-24h no estado de equilíbrio da estrona não conjugada (41%), da equilina (76%) e do 17-?-estradiol (22%). O efeito das doses de Etoricoxibe (substância ativa) recomendadas para uso crônico (60 mg e 90 mg) não foi estudado. Os efeitos da dose de Etoricoxibe (substância ativa) 120 mg na exposição (AUC0-24h) a esses componentes estrogênicos foram inferiores à metade daqueles observados quando o preparado de estrogênios conjugados foi administrado isoladamente e a dose foi aumentada de 0,625 mg para 1,25 mg. A importância clínica desse aumento é desconhecida e a administração de doses mais altas de estrogênios conjugados em combinação com Etoricoxibe (substância ativa) não foi estudada. Esse aumento na concentração estrogênica deve ser levado em consideração ao selecionar uma terapia de reposição hormonal para ser utilizada com Etoricoxibe (substância ativa).
Outros:
Em estudos de interação medicamentosa, Etoricoxibe (substância ativa) não exerceu efeitos clinicamente importantes na farmacocinética da prednisona/prednisolona ou da digoxina.
Antiácidos e Cetoconazol (um potente inibidor do CIP3A4):
Não exerceram efeitos clinicamente importantes na farmacocinética de Etoricoxibe (substância ativa).
Ação da Substância
Resultados da eficácia
Resultados dos estudos Artrite
Osteoartrite (OA)
Os pacientes com osteoartrite que receberam este medicamento apresentaram melhora significativa nas avaliações de dor, inflamação e mobilidade. Foram realizados dois estudos clínicos duplo-cegos, randômicos e com duração de até 52 semanas que envolveram aproximadamente 1.000 pacientes com agudização da OA no joelho ou no quadril; também foi avaliada OA da mão em 21% dos pacientes.
Nos dois estudos, a eficácia deste medicamento com 60 mg uma vez ao dia foi superior à do placebo durante um período de 12 semanas e comparável à de naproxeno 500 mg duas vezes ao dia durante todo o período de tratamento de 52 semanas. Os pacientes apresentaram redução significativa da dor e da rigidez articular, redução da sensibilidade articular causada pela dor e melhora significativa da mobilidade. A eficácia clínica foi demonstrada já no segundo dia de tratamento e manteve-se até o final dos estudos.
Nos pacientes com OA da mão, a redução da dor e da rigidez e a melhora da função física foram superiores às observadas com o placebo e semelhantes às observadas nos pacientes que receberam naproxeno.
Em um terceiro estudo, que envolveu aproximadamente 600 pacientes, a dose deste medicamento com 60 mg uma vez ao dia foi superior ao placebo durante um período de tratamento de seis semanas (no qual foram utilizadas avaliações semelhantes às dos dois primeiros estudos) e semelhante à de diclofenaco 50 mg três vezes ao dia na avaliação pelo paciente da resposta ao medicamento em estudo e na avaliação pelo pesquisador do status da doença durante um período de tratamento de até 92 semanas.
Artrite Reumatoide (AR)
Os pacientes com artrite reumatoide que receberam este medicamento apresentaram melhoras significativas nas múltiplas avaliações de dor, inflamação e mobilidade. Aproximadamente 1.700 pacientes com AR foram estudados em dois estudos clínicos duplo-cegos com 12 semanas de duração.
A dose deste medicamento 90 mg uma vez ao dia demonstrou eficácia superior à do placebo nos dois estudos. Em um estudo, este medicamento demonstrou eficácia semelhante à de naproxeno 500 mg duas vezes ao dia e, no outro estudo, eficácia superior à do naproxeno. Nesses dois estudos, os pacientes que receberam este medicamento apresentaram reduções clinicamente significativas no número de articulações dolorosas e de articulações edemaciadas e melhora nas avaliações da atividade da doença realizadas. A melhora com este medicamento também foi demonstrada pelo American College of Rheumatology 20% (ACR20) Responder Index, um composto de medidas clínicas, laboratoriais e funcionais de AR. Os efeitos benéficos deste medicamento foram observados logo após duas semanas (quando foi feita a primeira avaliação) e mantiveram-se até o final dos estudos.
Em um terceiro estudo que envolveu aproximadamente 600 pacientes e no qual foram utilizadas avaliações semelhantes às dos dois primeiros estudos, este medicamento com 90 mg uma vez ao dia demonstrou eficácia semelhante à do diclofenaco 50 mg três vezes ao dia durante um período de tratamento de 44 semanas.
Espondilite Anquilosante
Etoriceste medicamento xibe demonstrou melhorar de modo significativo a dor, a inflamação, a rigidez, a função e a mobilidade da coluna. este medicamento foi avaliado para o tratamento de espondilite anquilosante em um estudo clínico composto de duas partes, duplo-cego, de grupos paralelos, com duração de 52 semanas, que envolveu aproximadamente 400 pacientes. Na parte controlada com placebo, com duração de seis semanas, este medicamento na dose de 90 mg uma vez ao dia foi superior ao placebo em todos os desfechos primários (avaliação da dor na coluna pelo paciente, avaliação da atividade da doença pelo paciente e avaliação do índice funcional de espondilite anquilosante de Bath).
Além disso, na dose com 90 mg demonstrou efeitos estatisticamente superiores aos do naproxeno 500 mg duas vezes ao dia nas avaliações feitas pelos pacientes em relação à dor na coluna e à atividade da doença no período de seis semanas em que o estudo foi controlado com placebo. Os efeitos benéficos deste medicamento com 90 mg mantiveram-se durante o período de tratamento de 52 semanas, duplo-cego, com agente de comparação ativo. Os efeitos do tratamento com este medicamento 90 mg foram estatisticamente superiores em relação ao naproxeno nas avaliações de dor, inflamação, rigidez e função da coluna durante 1 ano. O benefício clínico deste medicamento foi observado logo após 4 horas do início do tratamento. Também foi estudada a dose de 120 mg deste medicamento uma vez ao dia, entretanto não foi observada eficácia adicional em comparação com a dose de 90 mg.
Dor Aguda, incluindo Dor Pós-Cirúrgica
Em um estudo de doses múltiplas pós-cirurgia odontológica, este medicamento com 90 mg administrado uma vez ao dia por até três dias proporcionou efeito analgésico significativamente maior em comparação com o placebo. Este medicamento 90 mg proporcionou tempo mais curto para início da ação e duração mais longa de alívio da dor, maior pico de alívio da dor, além de menor uso de analgésico de resgate após a dose inicial no primeiro dia em comparação com placebo. Este medicamento com 90 mg foi não inferior ao ibuprofeno 600 mg a cada 6 horas e foi superior ao paracetamol/codeína 600 mg/60 mg a cada 6 horas no alívio total da dor.
No estudo de histerectomia abdominal total, este medicamento com 90 mg foi administrado antes da cirurgia e por mais 4 dias9. As pacientes apresentaram intensidade de dor significativamente menor em repouso (média durante os 3 primeiros dias) em comparação com placebo.
Observou-se efeito benéfico nas primeiras 24 horas após a cirurgia, que se manteve durante o período de tratamento de 5 dias. As pacientes tratadas com este medicamento de 90 mg necessitaram de 30% menos morfina em média durante os 3 primeiros dias em comparação às pacientes do grupo placebo, resultando em recuperação mais rápida da motilidade intestinal.
Dor Crônica
Este medicamento aliviou a dor nos estudos de lombalgia crônica (aproximadamente 650 pacientes). O efeito analgésico deste medicamento foi demonstrado pela avaliação das respostas relacionadas à dor (por exemplo, sintomas, mobilidade e avaliações do tratamento pelos pacientes e pesquisadores). Este medicamento com 60 mg uma vez ao dia demonstrou eficácia significativa em uma semana de tratamento (quando foi feita a primeira avaliação) e a melhora da lombalgia crônica foi mantida nos pacientes que receberam este medicamento durante o período de 12 semanas de tratamento controlado com placebo.
Estudos Especiais
Programa de Estudo Multinacional deste medicamento (substância ativa deste medicamento) e Diclofenaco a Longo Prazo na Artrite (MEDAL)
O Programa MEDAL foi desenhado de forma prospectiva para avaliar resultados de segurança cardiovascular por meio dos dados combinados de três estudos individuais, randômicos, duplo-cegos e controlados com comparador ativo (diclofenaco) (estudo MEDAL, EDGE II e EDGE).
O programa MEDAL também avaliou a segurança no trato gastrintestinal superior e inferior. Esse programa incluiu 34.701 pacientes com osteoartrite e artrite reumatoide que receberam este medicamento 60 mg por dia (OA) ou este medicamento 90 mg por dia (OA e AR, 1,5 vez a dose recomendada para osteoartrite) versus diclofenaco 150 mg por dia por período médio de cerca de 18 meses; aproximadamente 12.800 pacientes tiveram mais que 24 meses de exposição ao tratamento e alguns, até 42 meses.
Os pacientes incluídos no programa apresentavam ampla gama de fatores de risco cardiovascular e gastrintestinal no período basal. Cerca de 47% dos pacientes tinham histórico de hipertensão, aproximadamente 12% dos pacientes tinham histórico de doença cardiovascular aterosclerótica (DCA) sintomática e cerca de 38% dos pacientes tinham, no período basal, risco cardiovascular elevado (definido como histórico anterior de doença cardiovascular aterosclerótica sintomática ou ? 2 fatores de risco cardiovascular dos 5 seguintes: tabagismo ou histórico de hipertensão, de diabetes mellitus, de dislipidemia ou de doença cardiovascular. Foram excluídos os pacientes com histórico recente de infarto do miocárdio, cirurgia de revascularização do miocárdio ou intervenção coronariana percutânea nos 6 meses anteriores à alocação no estudo. O uso de agentes gastroprotetores e de ácido acetilsalicílico em baixas doses foi permitido nos estudos; aproximadamente 50% dos pacientes utilizaram gastroprotetores e cerca de 35%, ácido acetilsalicílico em baixa dose. Nos estudos, a eficácia deste medicamento 60 mg e 90 mg demonstrou ser comparável à do diclofenaco.
Os dados de segurança cardiovascular e gastrintestinal estão resumidos abaixo.
Dados cardiovasculares:
O programa MEDAL demonstrou que a incidência de eventos adversos cardiovasculares trombóticos graves confirmados (que consistiram de eventos cardíacos, vasculares cerebrais e vasculares periféricos) foram comparáveis entre este medicamento e diclofenaco (veja tabela 1). Para o desfecho primário de eventos CV trombóticos confirmados, o risco relativo entre este medicamento e diclofenaco foi 0,95 (intervalo de confiança de 95%: 0,81; 1,11) na análise primária pré-especificada. A incidência para os tipos específicos de eventos trombóticos (por exemplo, infarto do miocárdio e acidente vascular cerebral) também foram semelhantes entre este medicamento e diclofenaco. A incidência foi semelhante entre este medicamento e diclofenaco ao longo de toda a duração do estudo, inclusive no subgrupo de pacientes que recebeu medicamento por mais de 24 meses. Não houve diferença significativa na incidência de eventos trombóticos entre este medicamento e diclofenaco em todos os subgrupos analisados, independente da categoria de risco cardiovascular dos pacientes no período basal. A mortalidade cardiovascular, assim como a mortalidade total, foram semelhantes entre os grupos deste medicamento e diclofenaco.
Tabela 1. Incidência de Eventos CV Trombóticos Confirmados (Dados Combinados do Programa MEDAL)
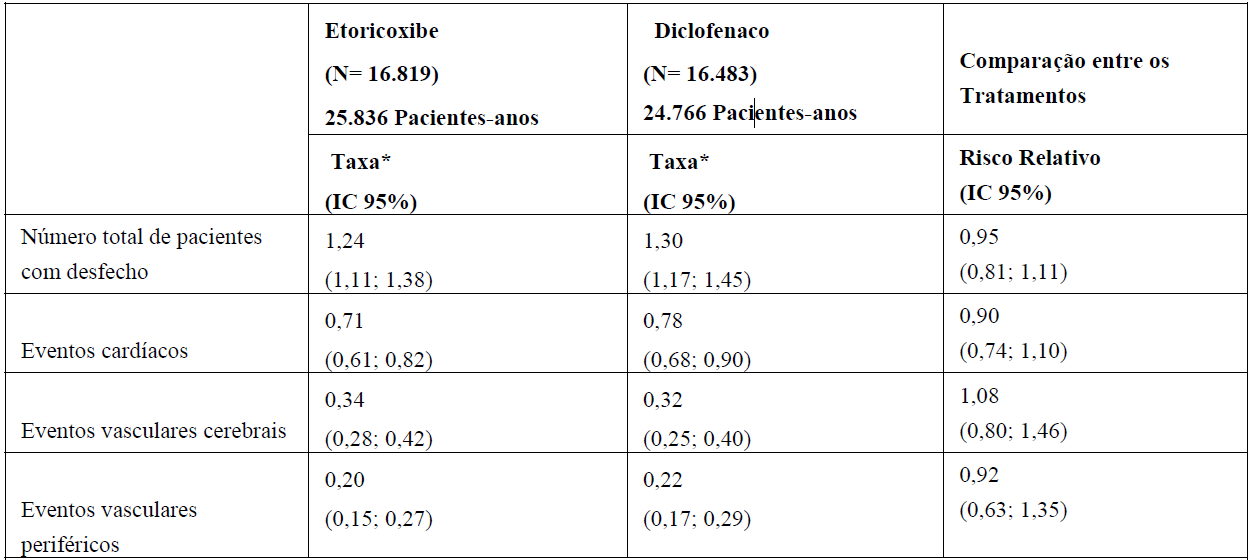
*Eventos por 100 pacientes-anos.
N= número total de pacientes; IC = intervalo de confiança.
Dados gastrintestinais:
A frequencia por 100 pacientes-anos de eventos clínicos confirmados do trato gastrintestinal superior (perfurações, úlceras e sangramentos - PUSs) foram 0,67 (IC 95%, 0,57; 0,77) com este medicamento e 0,97 (IC 95%, 0,85; 1,10) com diclofenaco, indicando risco relativo de 0,69 (IC 95%, 0,57; 0,83). A frequencia por 100 pacientes-anos de eventos clínicos complicados do trato GI superior foram semelhantes entre este medicamento e diclofenaco (0,30 vs. 0,32). Como o risco de eventos do trato GI superior aumenta com a idade, avaliou-se a frequencia desses eventos em pacientes idosos. A maior redução de risco foi observada em pacientes com idade ? 75 anos; a frequencia por 100 pacientes-anos para um evento confirmado no trato GI superior foi mais baixa para este medicamento do que para o diclofenaco (1,35 [IC 95%, 0,94; 1,87] vs. 2,78 [IC 95%, 2,14; 3,56]). Também foram avaliadas as taxas de eventos confirmados no trato GI superior para pacientes que receberam concomitantemente ácido acetilsalicílico em baixas doses e/ou agentes gastroprotetores; os dados estão na tabela 2.
A freqüência de eventos clínicos confirmados no trato GI inferior foram 0,32 (IC 95%, 0,25; 0,39) vs. 0,38 (IC 95%, 0,31; 0,46) por 100 pacientes-anos para este medicamento vs. diclofenaco, indicando risco relativo de 0,84 (IC 95%, 0,63; 1,13).
Tabela 2. Eventos Confirmados no Trato GI Superior (Dados Combinados do Programa MEDAL)
| Etoricoxibe (substância ativa) | Diclofenaco | |
| Frequencia* (IC 95%) | Frequencia* (IC 95%) | |
|
Frequencia total (Risco relativo 0,69 [0,57, 0,83]) | 0,67 (0,57; 0,77) | 0,97 (0,85; 1,10) |
| Uso concomitante de ácido acetilsalicílico em baixas doses | ||
| Não | 0,38 (0,29; 0,48) | 0,73 (0,60; 0,87) |
| Sim | 1,14 (0,94; 1,37) | 1,37 (1,15; 1,63) |
| Uso concomitante de agentes gastroprotetores** | ||
| Não | 0,63 (0,49; 0,79) | 0,83 (0,67; 1,02) |
| Sim | 0,70 (0,57; 0,84) | 1,07 (0,91; 1,25) |
*Frequencia = eventos por 100 pacientes-anos (PA) = (n/PA) x 100. IC = intervalo de confiança.
**Cerca de 96% dos agentes gastroprotetores utilizados corresponderam a inibidores.
Em cada estudo do programa MEDAL, também se avaliou a tolerabilidade gastrintestinal, definida como a ocorrência de descontinuações de pacientes do estudo por qualquer experiência adversa GI, seja clínica (por exemplo, dispepsia, dor abdominal e úlcera) ou laboratorial (por exemplo, aumento da ALT ou da AST), incluindo eventos hepáticos. O desfecho primário dos estudos EDGE e EDGE II foi a tolerabilidade gastrintestinal. Eles compararam este medicamento 90 mg por dia e diclofenaco 150 mg por dia em pacientes com osteoartrite (EDGE) e artrite reumatoide (EDGE II). Um dos objetivos secundários do estudo MEDAL foi comparar a tolerabilidade gastrintestinal entre este medicamento 60 mg (OA) ou 90 mg (OA e AR) e diclofenaco 150 mg por dia. Nos três estudos, este medicamento demonstrou tolerabilidade GI superior comparado ao diclofenaco (valores de p <0,001; veja figura 1). O benefício da tolerabilidade GI deste medicamento foi significativo para os componentes tanto clínicos quanto laboratoriais que compuseram esse desfecho.
Figura 1. Tolerabilidade Gastrintestinal
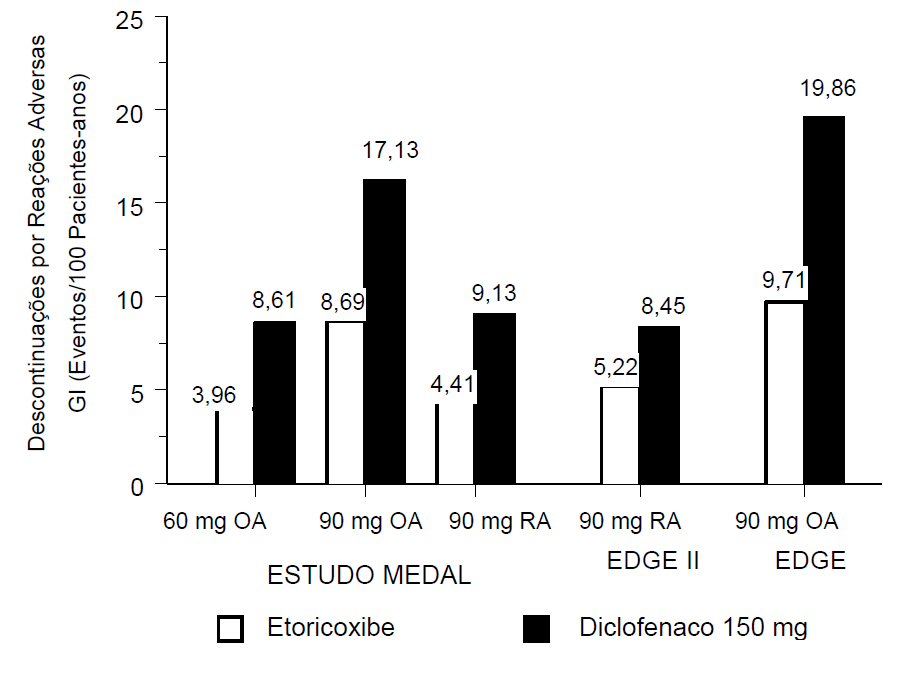
As reações adversas de origem hepática que levaram à descontinuação também foram avaliadas em cada estudo individual do programa MEDAL.
Nos três estudos, a incidência de descontinuação foi significativamente mais baixa nos grupos de tratamento com este medicamento 60 mg e 90 mg, em relação aos grupos com diclofenaco 150 mg, para pacientes com osteoartrite e artrite reumatoide.
Dados Adicionais de Segurança Cardiovascular Trombótica
Em uma análise combinada dos estudos clínicos de fase IIb a V com 4 semanas de duração ou mais (excluindo-se os estudos do Programa MEDAL), não houve diferença perceptível na taxa de eventos cardiovasculares trombóticos graves confirmados entre os pacientes que receberam este medicamento em doses ?30 mg (n= 2.147 pacientes; média de duração de exposição de aproximadamente 309 dias) ou AINEs, com exceção do naproxeno (ibuprofeno 2400 mg por dia ou diclofenaco 150 mg por dia, n= 1.470 pacientes; média de duração de exposição de aproximadamente 161 dias). A frequencia desses eventos foi mais alta em pacientes que receberam este medicamento (n= 1.960 pacientes; média de duração de exposição de aproximadamente 462 dias) em comparação com os que receberam naproxeno 500 mg duas vezes ao dia (n= 1.497 pacientes; média de duração de exposição de aproximadamente 421 dias). A diferença de atividade antiplaquetária entre alguns AINEs inibidores da COX-1 e inibidores seletivos da COX-2 pode ser de significância clínica em pacientes com risco de eventos tromboembólicos. Os inibidores seletivos da COX-2 reduzem a formação de prostaciclina sistêmica (e, portanto, possivelmente endotelial) sem afetar o tromboxano plaquetário.
Dados Adicionais de Segurança Gastrintestinal
Os estudos especiais a seguir foram conduzidos para avaliar se este medicamento, um inibidor seletivo da COX–2, está associado a menos toxicidade GI do que os AINEs não seletivos.
Endoscopia Digestiva Alta em Pacientes com Artrite Reumatoide ou Osteoartrite
A incidência cumulativa de úlceras gastroduodenais foi significativamente mais baixa em pacientes que receberam este medicamento 120 mg uma vez ao dia em comparação com os pacientes que receberam um de dois AINES não seletivos (naproxeno 500 mg duas vezes ao dia ou ibuprofeno 800 mg três vezes ao dia) em dois estudos endoscópicos, duplo-cegos, com duração de 12 semanas. No 1° estudo, foram envolvidos 700 pacientes com OA ou AR e no 2° estudo, 655 pacientes com OA. A incidência cumulativa de úlceras entre os pacientes que receberam este medicamento foi mais alta do que entre os que receberam placebo (ver os resultados desses estudos na figura 2).
Figura 2: Incidência Cumulativa de Úlcera Gastroduodenal ?3 mm* por Anos de Sobrevida após 12 Semanas para os Dois Estudos Endoscópicos (Intenção de Tratar)
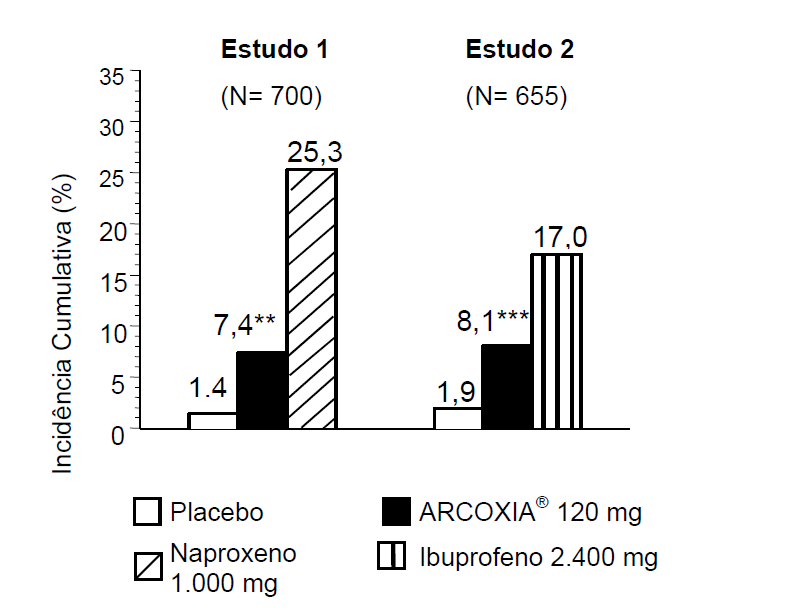
* Os resultados das análises cujo desfecho foi úlcera gastroduodenal ?5 mm foram compatíveis.
** p< 0,001 versus naproxeno 500 mg 2x/dia.
*** p= 0,007 versus ibuprofeno 800 mg 3x/dia.
Os dois estudos endoscópicos incluíram pacientes cujo risco de úlceras gastrintestinais (GI) era mais alto; isto é, pacientes com infecção ativa por Helicobacter pylori, com erosões gastroduodenais no período basal, histórico de perfuração, úlcera ou sangramento (PUSs) e/ou pacientes que faziam uso concomitante de corticosteroides. Dos pacientes incluídos, 400 (28%) tinham idade igual ou superior a 65 anos. A vantagem deste medicamento sobre o naproxeno ou o ibuprofeno foi mantida nesses subgrupos de risco mais alto.
Análise Combinada de Segurança Gastrintestinal
Em uma análise combinada de todos os estudos clínicos de fase IIb a V com 4 semanas de duração ou mais (excluindo-se os estudos do Programa MEDAL), a frequencia de eventos do tipo PUS (perfurações gastroduodenais, úlceras gastrintestinais sintomáticas ou sangramentos do trato GI superior) para doses combinadas deste medicamento variando entre 30 mg e 120 mg por dia (N= 4.107 pacientes; média de duração do tratamento de aproximadamente 220 dias) foi comparada à dos AINEs não seletivos (naproxeno 1.000 mg por dia, diclofenaco 150 mg por dia e ibuprofeno 2400 mg por dia; N total = 2.967 pacientes; média de duração do tratamento de aproximadamente 182 dias). A frequencia de eventos confirmados do tipo perfuração, úlcera ou sangramento no grupo deste medicamento foram cerca da metade das do grupo dos AINEs não seletivos, durante o primeiro ano de tratamento (1,13 eventos por 100 pacientes-anos para este medicamento e 2,64 eventos por 100 pacientes-anos para os AINEs; risco relativo 0,47 [IC 95%: 0,28, 0,76]). Os resultados foram consistentes ao longo de todo o período de acompanhamento.
Análise Combinada da Tolerabilidade Clínica Gastrintestinal
Uma análise combinada pré-especificada de oito estudos clínicos que envolveram aproximadamente 4.000 pacientes com OA, AR ou lombalgia crônica avaliou a incidência quanto aos seguintes desfechos: 1) descontinuação por sintomas no trato GI superior; 2) descontinuação por qualquer experiência adversa no trato GI; 3) pacientes que passaram a utilizar medicamentos gastroprotetores (incluindo antagonistas dos receptores H2, misoprostol e inibidores da bomba de prótons) e 4) pacientes que passaram a utilizar quaisquer medicamentos para o trato GI.
Houve redução de risco aproximada de 50% para esses desfechos entre os pacientes que receberam este medicamento (60 mg, 90 mg ou 120 mg uma vez ao dia) em comparação com os pacientes que receberam AINEs não seletivos (naproxeno 500 mg duas vezes ao dia ou diclofenaco 50 mg três vezes ao dia). Não houve diferenças estatisticamente significativas entre este medicamento e placebo.
Avaliação de Perda de Sangue Oculto nas Fezes em Indivíduos Sadios
Para avaliar a integridade da mucosa de todo o trato gastrintestinal, foram comparadas as perdas de sangue pelas fezes com a administração deste medicamento 120 mg uma vez ao dia, ibuprofeno 2.400 mg uma vez ao dia e placebo em um estudo que utilizou hemácias radiomarcadas com 51Cr e envolveu 62 homens sadios. Após quatro semanas de administração deste medicamento 120 mg, não houve aumento significativo na quantidade de perda de sangue nas fezes comparativamente ao placebo. Em contrapartida, a administração de ibuprofeno 2.400 mg/dia causou aumento significativo da perda de sangue fecal em comparação com os indivíduos que receberam placebo e os que receberam este medicamento.
Estudo da Função Renal em Pacientes Idosos
Um estudo com distribuição randômica, duplo-cego, controlado por placebo e de grupos paralelos avaliou os efeitos de 15 dias de tratamento deste medicamento (90 mg), celecoxibe (200 mg 2x/dia), naproxeno (500 mg 2x/dia) e placebo sobre excreção urinária de sódio, pressão arterial e outros parâmetros da função renal em pacientes com 60 a 85 anos de idade que recebiam uma dieta de 200 mEq/dia de sódio. Este medicamento, o celecoxibe e o naproxeno apresentaram efeitos similares sobre a excreção urinária de sódio durante as 2 semanas de tratamento. Todos os comparadores ativos apresentaram aumento na pressão arterial sistólica em relação ao placebo, no entanto este medicamento foi associado a aumento estatisticamente significativo no 14o dia em comparação com o celecoxibe e com o naproxeno (alteração média em relação ao período basal para pressão arterial sistólica: este medicamento 7,7 mmHg, celecoxibe 2,4 mmHg, naproxeno 3,6 mmHg).
Características farmacológicas
Mecanismo de Ação
Este medicamento é um anti-inflamatório não esteroide (AINE) que apresenta atividade anti-inflamatória, analgésica e antipirética em modelos animais.
Este medicamento é um potente inibidor da cicloxigenase-2 (COX-2), ativo por via oral, altamente seletivo, dentro e acima da faixa posológica clínica. Foram identificadas duas isoformas da cicloxigenase: a cicloxigenase-1 (COX-1) e a cicloxigenase-2 (COX-2). A COX-1 é responsável pelas funções fisiológicas normais mediadas pelas prostaglandinas, tais como a citoproteção gástrica e a agregação plaquetária. A inibição da COX-1 por anti-inflamatórios não esteroides (AINEs) não seletivos foi associada a lesões gástricas e redução da agregação plaquetária. A COX-2 demonstrou ser responsável principalmente pela síntese de mediadores prostanoides da dor, da inflamação e da febre. A inibição seletiva da COX-2 peleste medicamento diminui esses sinais e sintomas clínicos com menos toxicidade gastrintestinal e sem exercer efeito na função plaquetária.
Em estudos de farmacologia clínica, este medicamento produziu inibição de COX-2 dose-dependente sem inibição da COX-1, em doses de até 150 mg/dia.
A influência sobre a atividade gastroprotetora da COX-1 também foi avaliada em um estudo clínico no qual a síntese de prostaglandinas foi medida em amostras de biópsias gástricas de indivíduos que receberam 120 mg deste medicamento diariamente, 500 mg de naproxeno duas vezes ao dia ou placebo. Este medicamento não inibiu a síntese gástrica de prostaglandina comparativamente ao placebo; em contrapartida, o naproxeno inibiu a síntese gástrica de prostaglandina em aproximadamente 80% quando comparado ao placebo. Esses dados reforçam a seletividade deste medicamento para a COX-2.
Função Plaquetária
Múltiplas doses deste medicamento de até 150 mg/dia administradas por até nove dias não exerceram efeito no tempo de sangramento em relação ao placebo. De forma similar, o tempo de sangramento não foi alterado em um estudo com administração de dose única de 250 mg ou 500 mg deste medicamento. Não houve inibição, no estado de equilíbrio, da agregação plaquetária ex vivo induzida pelo ácido araquidônico ou por colágeno com doses de até 150 mg de etorioxibe. Esses achados são compatíveis com a seletividade deste medicamento para a COX-2.
Absorção
Este medicamento é bem absorvido por via oral. A biodisponibilidade oral média é de aproximadamente 100%. Em adultos, em jejum, o pico de concentração plasmática (média geométrica da Cmáx= 3,6 ?g/mL) foi observado aproximadamente 1 hora (Tmáx) após a administração de 120 mg uma vez ao dia até o estado de equilíbrio. A média geométrica da área sob a curva (AUC0-24h) foi de 37,8 ?g•h/mL. A farmacocinética deste medicamento é linear na faixa posológica clínica.
Uma refeição-padrão não exerceu efeito clinicamente significativo na magnitude ou velocidade de absorção de uma dose de 120 mg deste medicamento; em estudos clínicos, este medicamento foi administrado independentemente da alimentação. A farmacocinética deste medicamento foi semelhante (AUC comparável, Cmáx em aproximadamente 20%) quando administrado isoladamente ou com antiácidos contendo hidróxido de alumínio/magnésio ou carbonato de cálcio (aproximadamente 50 mEq de capacidade neutralizadora do ácido) a 12 indivíduos sadios.
Distribuição
A ligação às proteínas plasmáticas humanas deste medicamento é de aproximadamente 92% na faixa de concentração de 0,05 a 5 ?g/mL. O volume de distribuição no estado de equilíbrio (Vdss) é de aproximadamente 120 litros em seres humanos. Este medicamento atravessa a placenta de ratas e coelhas e a barreira hematoencefálica de ratos.
Metabolismo
Este medicamento é amplamente metabolizado e menos de 1% da dose é recuperada na urina de forma inalterada. A principal via metabólica para a formação do metabólito 6'-hidroximetil é catalisada pelas enzimas do citocromo P450 (CIP).
Cinco metabólitos foram identificados em humanos; o principal é o 6'-ácido carboxílico derivado deste medicamento formado pela oxidação adicional do metabólito 6'-hidroximetil. Esses metabólitos principais ou não demonstram nenhuma atividade mensurável ou apresentam apenas fraca atividade como inibidores da COX-2; nenhum deles inibe a COX-1.
Eliminação
Após administração intravenosa de uma dose de 25 mg deste medicamento, marcada radioativamente, a indivíduos sadios, 70% da radioatividade foi recuperada na urina e 20%, nas fezes, a maioria como metabólito; menos de 2% foram recuperados como fármaco inalterado.
A eliminação deste medicamento é feita quase exclusivamente pelo metabolismo, seguida de excreção renal. Concentrações de estado de equilíbrio deste medicamento são atingidas 7 dias após a administração de 120 mg uma vez ao dia, com relação de acúmulo de aproximadamente 2, correspondente à meia-vida de acúmulo de aproximadamente 22 horas. O clearance plasmático é estimado em aproximadamente 50 mL/min.
Cuidados de Armazenamento
Conservar em temperatura ambiente (entre 15 e 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aparência:
- Hetori 60 mg: comprimido revestido verde-escuro, biconvexo, em forma de maçã, com a inscrição “200” em um lado.
- Hetori 90 mg: comprimido revestido branco, biconvexo, em forma de maçã, com a inscrição “202” em um lado.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
Venda sob prescrição médica.
Só pode ser vendido com retenção da receita.
MS 1.0171.0200
Farm. Resp.:
Cristina Matushima - CRF-SP no 35.496
Registrado e importado por:
Schering-Plough Indústria Farmacêutica Ltda.
Rua João Alfredo, 353 – São Paulo/SP
CNPJ 03.560.974/0001-18 – Indústria Brasileira
SAC Grünenthal: 0800 205 2050
Fabricado por:
Frosst Ibérica S.A.
Madri, Espanha
Embalado por:
Merck Sharp & Dohme Farmacêutica Ltda.
Rua 13 de Maio, 1.161 - Sousas, Campinas/SP
Comercializado por:
Grünenthal do Brasil Farmacêutica Ltda.
Av. Guido Caloi, 1935, BL B e BL C - 1o andar
São Paulo/SP