Granulokine 300mcg, caixa com 1 seringa preenchida com 0,5mL de solução de uso subcutâneo ou intravenoso na Pharmed
Granulokine - 30 Mu 300 Mcg Solução Injetável 1 Seringa Preenchida 0,5 Ml

Granulokine 300mcg, caixa com 1 seringa preenchida com 0,5mL de solução de uso subcutâneo ou intravenoso na Pharmed
Não encontramos este produto para a Pharmed no Cliquefarma.
Confira os menores preços para Granulokine - 30 Mu 300 Mcg Solução Injetável 1 Seringa Preenchida 0,5 Ml
Granulokine - 30 Mu 300 Mcg Solução Injetável 1 Seringa Preenchida 0,5 Ml
54.91%
R$ 230,55
Mundial Farma
51.18%
R$ 249,60
Onco Express Medicamentos Especiais e Oncológicos
47.87%
R$ 266,50
Saúde Farma Medicamentos
46.99%
R$ 271,00
OncoExpresso Medicamentos
43.47%
R$ 289,00
Para que serve
Granulokine é indicado para redução da duração da neutropenia (diminuição do número de glóbulos brancos neutrófilos) e da incidência da neutropenia febril em pacientes com neoplasias (tumores) não originadas da medula óssea, tratados com quimioterapia citotóxica estabelecida (medicamentos que atacam as células).
Granulokine também é indicado para redução da duração da neutropenia e suas sequelas clínicas em pacientes submetidos à terapia mieloablativa (que destrói a medula óssea) seguida de transplante de medula óssea.
Peça ao médico mais esclarecimentos sobre a sua doença se necessário.
Como Granulokine funciona?
Granulokine é uma proteína altamente purificada produzida em laboratório que age estimulando especificamente a produção de neutrófilos (um dos tipos de glóbulos brancos que também são chamados de granulócitos), que ajudam o organismo a se defender contra infecções, principalmente bacterianas.
Em 24 horas após administração, ocorre aumento evidente dessas células.
A redução do número de granulócitos, que são produzidos na medula óssea, pode ocorrer nas seguintes situações:
Com o uso de quimioterapias tóxicas para a medula óssea, em infecções graves ou em outras situações.
Contraindicação
Granulokine não deverá ser administrado caso você tenha alergia ao filgrastim ou aos demais componentes.
Granulokine não deve ser administrado para aumentar a dose de quimioterapia contra câncer acima dos esquemas de administração e doses já estabelecidos pelo seu médico.
Você não deve receber Granulokine se for portador de neutropenia congênita severa (Síndrome de Kostmann) com constituição genética anormal.
Como usar
Uma ou duas doses de Granulokine por dia devem ser aplicadas por via subcutânea ou intravenosa. A aplicação de Granulokine deve ser feita ou supervisionada por um médico com experiência no uso desse tipo de medicamento.
Quimioterapia citotóxica estabelecida
A dose recomendada de Granulokine é de 0,5 MU/kg/dia (5 mcg/kg/dia). O conteúdo de uma seringa ou um único frasco de Granulokine com 30 milhões de unidades fornece, portanto, a dose diária necessária para um paciente com 60 kg. A primeira dose de Granulokine não deve ser administrada em menos de 24 horas após a quimioterapia citotóxica.
Espera-se que a duração necessária do tratamento para que o número dos neutrófilos retorne aos valores normais seja de até 14 dias, dependendo do tipo, da dose e do esquema quimioterápico citotóxico utilizado.
Em pacientes sob quimioterapia citotóxica, uma elevação transitória do número de neutrófilos é tipicamente observada 1 a 2 dias após iniciado o tratamento com Granulokine. Contudo, é recomendado manter a aplicação diária até que você recupere os valores normais do número de neutrófilos.
Pacientes tratados com terapia mieloablativa seguida de transplante da medula óssea
A dose inicial recomendada de Granulokine é de 1,0 MU/kg/dia (1 milhão de unidades por quilo de peso por dia) (10 mcg/kg/dia) (10 microgramas por quilo de peso por dia) administrado em 30 minutos ou 24 horas por infusão intravenosa (na veia), ou 1,0 MU/kg/dia (10 mcg/kg/dia) (mesma dose) administrado em 24 horas, de maneira contínua, por via subcutânea (abaixo da pele).
Granulokine deve ser diluído em 20 mL de solução glicosada a 5% (soro glicosado 5%).
A primeira dose de Granulokine não deve ser administrada nas 24 horas seguintes à quimioterapia citotóxica, mas sim dentro das 24 horas após a infusão da medula óssea. A eficácia e a segurança da administração de Granulokine por mais 28 dias neste contexto ainda não foram estabelecidas.
Granulokine tem demonstrado eficácia e boa tolerabilidade neste contexto em doses até 70 mcg/kg/dia.
Uso em idosos
Estudos clínicos com Granulokine incluíram pequeno número de pacientes idosos. Estudos especiais não foram realizados nesse grupo e, portanto, recomendações específicas de dosagem não podem ser feitas.
Insuficiência renal ou hepática
Não foram realizados estudos com Granulokine em pacientes com prejuízo severo das funções hepática e renal.
Portanto, seu uso nestes pacientes não pode ser recomendado.
O profissional da saúde saberá como preparar o medicamento.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que fazer quando eu me esquecer de usar Granulokine?
Seu médico saberá quando deverá ser aplicada a próxima dose de Granulokine.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Geral
Em pacientes tratados com filgrastim, foi reportada alergia, incluindo reações anafiláticas (um tipo de reação alérgica grave), que ocorrem no início ou durante o tratamento.
Os pacientes com alergia clinicamente significativa devem parar permanentemente o tratamento com filgrastim.
Os pacientes com histórico de alergia ao filgrastim ou ao pegfilgrastim não devem utilizá-lo.
Existem trabalhos publicados na literatura médica relatando que um número elevado de glóbulos brancos no sangue pode piorar a evolução de pacientes com anemia falciforme. Se você é portador de anemia falciforme, precisa informar ao seu médico, pois serão necessários exames clínicos e laboratoriais adicionais.
É possível que o Granulokine provoque aumento das crises de falcização.
A contagem de plaquetas deve ser cuidadosamente monitorada, pois trombocitopenia (redução do número de plaquetas no sangue) foi frequentemente relatada em pacientes em tratamento com Granulokine.
Se você é portador de osteoporose ou osteopenia, poderá haver necessidade de acompanhamento com exames periódicos de densitometria se você for tratado com Granulokine durante mais do que 6 meses.
Se você já foi submetido a radio e/ou quimioterapia extensas ou apresenta infiltração da medula óssea pelo tumor, pode ser que já exista diminuição das células da medula responsáveis pela produção de neutrófilos, os progenitores mieloides.
Se isso ocorrer, o aumento de neutrófilos com o uso do Granulokine poderá não ser o esperado.
O volume do baço deve ser avaliado regularmente por meio de palpação abdominal ou ultrassom para detecção de aumentos anormais.
Granulokine contém sorbitol como excipiente numa concentração de 50 mg/mL. É improvável que o tratamento apenas com Granulokine resulte em toxicidade clinicamente relevante para indivíduos sensíveis.
No entanto, em casos de intolerância hereditária à frutose, aconselha-se cautela.
Ainda não se conhece o efeito de Granulokine sobre a doença do enxerto versus hospedeiro (GvHD).
Início de febre ou sinais pulmonares (como tosse, falta de ar) em associação a sinais radiológicos de infiltrados pulmonares e piora da função pulmonar, pode corresponder a sinais preliminares da síndrome da angústia respiratória aguda (SARA), um quadro pulmonar grave. Em tais circunstâncias, seu médico deverá recomendar que você pare de usar Granulokine, e o tratamento apropriado para essa síndrome deve ser iniciado.
Glomerulonefrite (inflamação nos rins) tem sido relatada em pacientes que receberam tratamento com filgrastim e pegfilgrastim.
Geralmente, eventos de glomerulonefrite são resolvidos após redução da dose ou descontinuação do uso de filgrastim e pegfilgrastim. O monitoramento através de exames de urina deve ser feito conforme orientação médica.
Crescimento de células malignas
Granulokine pode promover o crescimento de células mieloides (células da medula óssea) in vitro, e efeitos semelhantes podem ser observados em algumas células não mieloides in vitro.
A segurança e a eficácia da administração do Granulokine em pacientes com síndrome mielodisplásica, leucemia mieloide aguda (LMA) ou leucemia mieloide crônica (LMC) não foram estabelecidas. Portanto, devido à possibilidade de crescimento do tumor, Granulokine deve ser administrado com extrema cautela nesses casos.
Em vista de dados de eficácia e segurança limitados em pacientes com leucemia meloide aguda secundária, Granulokine deve ser administrado com cautela.
Em pacientes recebendo quimioterapia citotóxica (que destrói as células)
Aumento do número de leucócitos (glóbulos brancos)
Como pode ocorrer aumento do número de glóbulos brancos no sangue, e isso pode gerar riscos para você, deve-se realizar contagem das células frequentemente durante a terapia com Granulokine. Menos de 5% dos pacientes recebendo doses elevadas de Granulokine (equivalentes a 10 seringas por dia) chegaram a apresentar numero de glóbulos brancos ? 100 x 109/L e, mesmo nesses casos, não apareceram efeitos colaterais decorrentes desse aumento.
Mas como existem alguns riscos com o aumento excessivo do numero de glóbulos brancos, é necessário fazer hemogramas (exame de sangue) com contagem dos leucócitos periodicamente durante o tratamento. Se o número de glóbulos brancos ultrapassar 50 x 109/L logo depois de ter atingido o valor mais baixo, Granulokine deve ser descontinuado imediatamente.
Riscos associados a altas doses de quimioterapia
Granulokine pode possibilitar que você receba o esquema de quimioterapia indicado, sem que os glóbulos brancos cheguem a um nível tão baixo que seja necessário suspender o tratamento. No entanto, ele não deve ser utilizado para permitir o uso de doses ou frequência de quimioterapia maiores do que as indicadas no seu caso, porque não está comprovado que essas doses elevadas tenham melhor resultado sobre as células do tumor e também podem provocar aumento de toxicidade, com efeitos indesejáveis sobre coração, pulmões, sistema nervoso e pele.
Granulokine não age sobre a redução de outros elementos do sangue, como glóbulos vermelhos e plaquetas, que podem apresentar redução com a quimioterapia.
Como o tratamento com Granulokine pode permitir que você receba o esquema de quimioterapia com doses mais adequadas ao combate do tumor sem apresentar queda tão acentuada dos glóbulos brancos, será necessário acompanhar cuidadosamente os exames de sangue periódicos para verificar os níveis de plaquetas e glóbulos vermelhos, principalmente se os quimioterápicos indicados no seu caso tiverem mais efeito sobre as plaquetas.
Imunogenicidade
Como em qualquer tratamento com medicamentos compostos por proteínas, existe um potencial de desenvolvimento de imunogenicidade (ativar o sistema imune) durante o uso de filgrastim. No entanto, as taxas de geração de anticorpos contra essa substância são geralmente baixas e não houve consequências clínicas adversas.
Efeitos sobre a capacidade de dirigir e operar máquinas
Não foram relatados efeitos sobre a capacidade de dirigir e operar máquinas.
Até o momento, não há informações de que Granulokine (filgrastim) possa causar doping. Em caso de dúvidas, consulte o seu médico.
Gravidez e amamentação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
A segurança do Granulokine não foi estabelecida em mulheres grávidas. Existem trabalhos na literatura médica demonstrando que pode haver passagem de filgrastim da gestante para o feto através da placenta.
Estudos em animais de laboratório mostraram que pode haver toxicidade para a reprodução.
Durante a gestação, seu médico deverá avaliar o possível risco do uso de Granulokine para o feto, com relação aos benefícios terapêuticos esperados.
Não se sabe se o Granulokine passa para o leite materno. Granulokine não é recomendado para mulheres que estejam amamentando.
Não foram estabelecidas a segurança e a eficácia de Granulokine em crianças.
Testes laboratoriais
É recomendado o monitoramento do hemograma completo durante o tratamento com filgrastim.
Interações medicamentosas
A segurança e a eficácia do Granulokine administrado no mesmo dia da quimioterapia tóxica para a medula óssea não foram estabelecidas.
O uso de Granulokine não é recomendado no período de 24 horas antes até 24 horas após quimioterapia.
Evidências preliminares em um número pequeno de pacientes tratados concomitantemente com Granulokine e 5-fluorouracil indicam que a gravidade da neutropenia pode ser exacerbada.
Embora a interação com lítio, que também promove a liberação de neutrófilos, não tenha sido formalmente investigada, não há evidências de que seja prejudicial.
Possíveis interações com outros fatores de crescimento hematopoiéticos e citocinas ainda não foram investigadas.
Imagem óssea
A atividade aumentada da medula óssea para produção de células sanguíneas em resposta à terapia com fator de crescimento tem sido associada a alterações temporárias de imagens ósseas, o que deve ser considerado na interpretação dos resultados de exames de imagem.
Pode ocorrer alteração temporária de imagens ósseas em exames de imagem associada ao uso de Granulokine.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Em pacientes com câncer
Reação muito comum (ocorre em 10% ou mais dos pacientes que recebem esse medicamento):
Náusea, vômito, aumento da gama-glutamil transferase (GGT), aumento da fosfatase alcalina, aumento da desidrogenase lática (DHL) e do ácido úrico.
Reação comum (ocorre entre 1% e 10% dos pacientes que recebem esse medicamento):
Fadiga, fraqueza generalizada, inflamação da mucosa, dor de cabeça, prisão de ventre, diarreia, anorexia (redução ou ausência de apetite), dor no peito, dor musculoesquelética, tosse, dor na garganta, alopécia (queda de pelo e cabelo) e erupção cutânea.
Reação incomum (ocorre entre 0,1% e 1% dos pacientes que recebem esse medicamento):
Dor inespecífica.
Reação muito rara (ocorre em menos 0,01% dos pacientes que recebem esse medicamento):
Reações alérgicas, piora da artrite reumatoide, infiltrados pulmonares, Síndrome de Sweet (dermatose com placas vermelhas salientes na face, no pescoço e nos membros), vasculites (inflamação dos vasos) cutâneas e anormalidades na urina.
Dor nos ossos e dor nas extremidades ocorreram com uma incidência alta em pacientes tratados com filgrastim, quando comparado com pacientes tratados com placebo, em todas as indicações.
Experiência pós-comercialização
Distúrbios do sistema imunológico
Reações alérgicas incluindo anafilaxia (reação alérgica grave, acompanhada de queda acentuada da pressão arterial e dificuldade para respirar), rash cutâneo (erupção na pele) e urticária (reação alérgica em que aparecem placas avermelhadas, com muita coceira), podem ocorrer no tratamento inicial ou subsequente em pacientes recebendo filgrastim.
Em alguns casos, os sintomas ocorreram novamente quando o paciente recebeu uma nova dose de filgrastim, sugerindo uma relação de causa entre medicamento e efeito.
Reações alérgicas a filgrastim foram raramente reportadas em experiência pós-comercialização. Caso você apresente alguma reação alérgica séria, o seu médico poderá descontinuar o uso de Granulokine.
Distúrbios do sangue e do sistema linfático
Casos isolados de crise de falcização (hemácias perdem a forma normal e assumem a forma de “foice”), em alguns casos fatais, foram relatados em pacientes com anemia falciforme.
Casos frequentes de esplenomegalia (aumento do volume do baço) foram relatados em pacientes tratados com Granulokine (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento).
Casos pouco frequentes de ruptura de baço foram relatados em doadores normais e pacientes recebendo G-CSFs (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento).
Distúrbios musculoesqueléticos
Eventos de pseudogota foram relatados muito raramente em pacientes com câncer tratados com Granulokine (0,00003%).
Distúrbios de pele e tecidos subcutâneos
Foram relatados casos raros (ocorre entre 0,01% e 0,1% dos pacientes que utilizam este medicamento) de Síndrome de Sweet (dermatite febril aguda).
Reações de vasculite cutânea foram relatadas muito raramente em pacientes com câncer que receberam Granulokine (0,001%).
Distúrbios renais e urinários
Foram relatados casos de glomerulonefrite (inflamação nos rins).
Anormalidades laboratoriais
Elevações leves a moderadas e reversíveis de ácido úrico, fosfatase alcalina e desidrogenase láctica, sem associação com efeitos clínicos, foram observadas em pacientes recebendo filgrastim após quimoterapia
citotóxica.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através de seu serviço de atendimento.
Composição
Cada frasco-ampola de 1 mL ou seringa preenchida de 0,5 mL contém:
Filgrastim - 30 milhões de unidades (MU) – 300 mcg.
Excipientes: Hidróxido de sódio, ácido acético glacial, sorbitol, polissorbato 80 e água para injeção.
Filgrastim é produzida em cepa de laboratório de Escherichia coli manipulada geneticamente mediante inclusão de um gene para o fator estimulador de colônias de granulócitos.
Superdosagem
Não foram estabelecidos os efeitos de Granulokine em caso de superdosagem. Doses de até 138 mcg/kg/dia foram administradas sem efeitos tóxicos. A descontinuação da terapia com Granulokine,em geral, resulta na diminuição de 50% dos neutrófilos circulantes dentro de um a dois dias, com retorno aos níveis normais em um a sete dias.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Não foram ainda estabelecidas a segurança e a eficácia de Filgrastim (substância ativa) quando administrado no mesmo dia que a quimioterapia citotóxica mielossupressora. Tendo em vista a sensibilidade da rapidez da divisão das células mieloides à quimioterapia citotóxica mielossupressora, a utilização de Filgrastim (substância ativa) não é recomendada no período que decorre entre as 24 horas anteriores e às 24 horas posteriores à quimioterapia. Evidências preliminares obtidas a partir de um grupo pequeno de pacientes tratados concomitantemente com Filgrastim (substância ativa) e 5-fluoruracila indicam que a gravidade da neutropenia pode ser exacerbada.
Nos estudos clínicos conduzidos não foram investigadas as possíveis interações do Filgrastim (substância ativa) com outros fatores de crescimento hematopoiéticos e com citocinas.
Considerando que o lítio promove a liberação de neutrófilos, é provável que possa potencializar o efeito de Filgrastim (substância ativa). No entanto, esta interação não foi formalmente investigada e não existe qualquer evidência de que tal interação possa ser prejudicial.
Ação da Substância
Resultados de eficácia
A eficácia e segurança clínica do Filgrastim (substância ativa) foram avaliadas em três estudos clínicos fase III.
Câncer de mama
Estudo clínico fase III, multinacional, multicêntrico, randomizado e controlado de comparação entre Filgrastim (substância ativa) vs. comparador vs. placebo, conduzido em 348 pacientes com câncer de mama tratados com quimioterapia citotóxica. Durante o estudo, os voluntários estavam sob tratamento quimioterápico de no máximo 4 ciclos de docetaxel 75mg/m2 IV e doxorrubicina 60mg/m2 IV no 1° dia.
Os resultados de eficácia basearam-se nas seguintes determinações:
Duração de neutropenia severa (DSN): a duração de neutropenia severa no ciclo 1 foi de 1,1 dias (faixa de 0 a 5) para pacientes tratados com o Filgrastim (substância ativa) e o medicamento comparador e 3,8 dias (faixa de 0 a 9) em pacientes que utilizaram o placebo.
Os resultados foram similares no total de pacientes tratados, confirmando a comparabilidade de Filgrastim (substância ativa) e o medicamento comparador.
Incidência de neutropenia febril (FN): no ciclo 1, a incidência de neutropenia febril observada ou definida em protocolo foi consideravelmente menor nos grupos tratados com Filgrastim (substância ativa) e com o medicamento comparador, comparado aos grupos que receberam o placebo (12,1% vs. 12,5% vs. 36,1%). Não houve diferenças significativas entre o Filgrastim (substância ativa) e o medicamento comparador na incidência de neutropenia febril no ciclo 1 ou nos demais ciclos.
Contagem absoluta de neutrófilos (ANC): no ciclo 1, grupos que receberam Filgrastim (substância ativa) e o medicamento comparador apresentaram um aumento significativo de ANC após o 2° dia de tratamento, atingindo contagem máxima no 3° dia de tratamento.
Em seguida, os grupos demonstraram diminuição de ANC a 0,7 x 109/L no 7° dia e alcançaram novamente uma contagem máxima no 11° dia. No grupo que recebeu placebo, não houve aumento inicial de ANC, sendo observada diminuição constante a partir do 2° dia, atingindo um nível consideravelmente baixo (0,2 x 109/L) no 11° dia.
No ciclo 1, o tempo médio de recuperação na contagem absoluta de neutrófilos foi similar nos grupos de que receberam o Filgrastim (substância ativa) e o medicamento comparador (8 dias) e consideravelmente maior no grupo que recebeu placebo (15 dias).
Nos ciclos 2 a 4, o ANC foi similar para todos os grupos (~1,0 x 109/L) e o tempo médio de recuperação na contagem absoluta de neutrófilos foi de 8 dias para todos os grupos.
Neste estudo clínico fase III, conduzido com pacientes de alto-risco ou com câncer de mama avançado, Filgrastim (substância ativa) demonstrou ser superior ao placebo, e com eficácia comparável à do medicamento comparador na redução da duração de neutropenia severa induzida pela quimioterapia, no aumento da contagem absoluta de neutrófilos e na redução do tempo de recuperação da contagem absoluta de neutrófilos. O Filgrastim (substância ativa) e o medicamento comparador também demonstraram eficácia equivalente na redução da incidência de neutropenia febril quando comparados ao placebo.
Os resultados de eficácia deste estudo são sumarizados na tabela a seguir: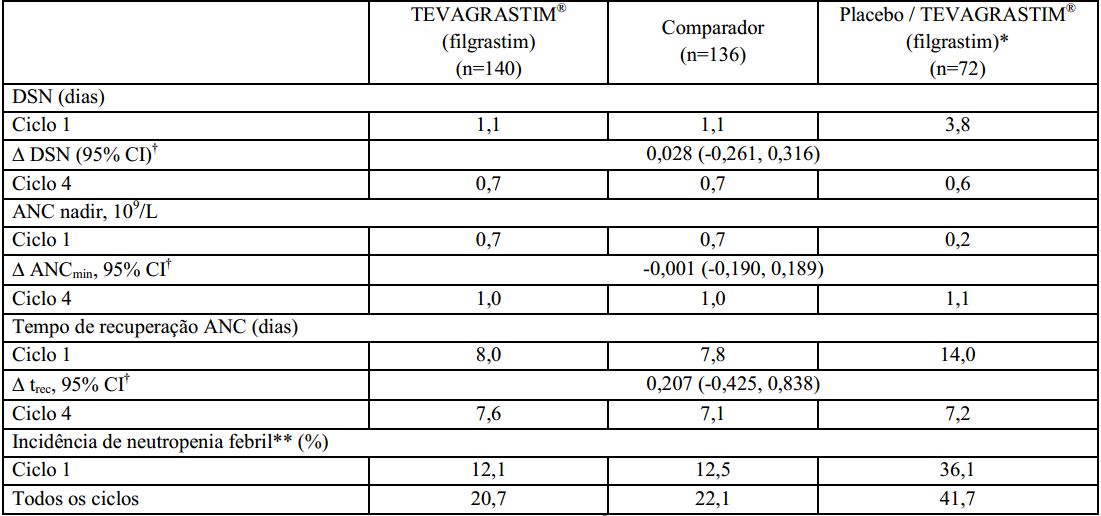 *Pacientes deste grupo receberam placebo no ciclo 1 e Filgrastim (substância ativa) nos ciclos de 2 a 4;
*Pacientes deste grupo receberam placebo no ciclo 1 e Filgrastim (substância ativa) nos ciclos de 2 a 4;
† Estimativa de análise de covariância (ANCOVA) e o intervalo de confiança de 95% para a diferença entre o Filgrastim (substância ativa) e o medicamento comparador no ciclo 1;
**Observado ou definido em protocolo;
DSN: Duração de neutropenia severa;
ANC: Contagem absoluta de neutrófilos;
trec: Tempo para recuperação da contagem absoluta de neutrófilos;
FN: Incidência de neutropenia febril.
Câncer de pulmão
Estudo clínico fase III, multinacional, multicêntrico e randomizado, de comparação entre Filgrastim (substância ativa) e medicamento comparador, conduzido em 240 pacientes com câncer de pulmão tratados com quimioterapia citotóxica. Durante o estudo, os voluntários estavam sob tratamento quimioterápico de no máximo 6 ciclos com derivados de platina. O regime de terapia mais comum utilizado foi cisplatina + etoposídeo ou gencitabina em 49% e 15% dos pacientes, respectivamente. Outros regimes incluíram cisplatina + vinorelbina e combinações de carboplatina + vinorelbina, etoposídeo, gencitabina ou paclitaxel.
Os resultados de eficácia basearam-se nas seguintes determinações:
Duração de neutropenia severa (DSN): a duração de neutropenia severa no ciclo 1 foi de 0,5 dias para pacientes tratados com o Filgrastim (substância ativa) e 0,3 dias para os pacientes tratados com o medicamento comparador. A estimativa de análise de covariância (ANCOVA) entre o Filgrastim (substância ativa) e o medicamento comparador foi de 0,157 dias. O intervalo de confiança de 95% (-0,114 a 0,428 dias) foi incluído na faixa pré-definida de equivalência (-1 a 1 dia), indicando que a duração de neutropenia severa (DSN) não foi diferente para o Filgrastim (substância ativa) e o medicamento comparador.
Contagem absoluta de neutrófilos (ANC): o perfil de contagem absoluta de neutrófilos foi similar em todos os ciclos para o medicamento comparador e o Filgrastim (substância ativa). Houve um aumento inicial de ANC significativo atingindo contagem máxima no 5° dia e subsequente diminuição nos 11° e 12° dias. ANC atingiu uma segunda contagem máxima no 14° dia e retornou próxima a contagem basal, gradualmente, até o 21° dia. A contagem absoluta de neutrófilos foi comparável entre os grupos que receberam o medicamento comparador e Filgrastim (substância ativa) no ciclo 1 (2,1 vs. 2,9 x 109/L) e após migrarem do medicamento comparador para Filgrastim (substância ativa) no ciclo 4 (2,3 vs. 3,2 x 109/L). No tempo médio de recuperação na contagem absoluta de neutrófilos houve diferenças mínimas entre os grupos no ciclo 1 (6,3 vs. 4,5 dias) que persistiu até o ciclo 4 quando Filgrastim (substância ativa) foi administrado em ambos os grupos (6,4 vs. 4,5 dias).
Incidência de neutropenia febril (FN):
No ciclo 1, a incidência observada ou definida em protocolo foi de 15,0% no grupo que recebeu o Filgrastim (substância ativa) e 8,8% no grupo que recebeu o medicamento comparador. Estatisticamente, esta diferença não é significativa (p=0,23). No ciclo 4, após os pacientes migrarem do medicamento comparador para Filgrastim (substância ativa), a incidência de neutropenia febril foi de 4,3% e 3,3%, respectivamente (p=0.90). Nos outros ciclos, a incidência de neutropenia febril foi de 33,1% e 23,8% nos pacientes sob tratamento com o Filgrastim (substância ativa) e o medicamento comparador, respectivamente.
Neste estudo clínico fase III, conduzido com pacientes com câncer de pulmão sob tratamento quimioterápico citotóxico, a profilaxia primária com Filgrastim (substância ativa) e o medicamento comparador demonstraram eficácia e segurança equivalentes. O perfil de contagem absoluta de neutrófilos, incluindo a duração de neutropenia severa foi similar entre ambos os medicamentos. Diferenças estatisticamente não significativas entre o Filgrastim (substância ativa) e o medicamento comparador na incidência de neutropenia febril podem ser atribuídas às diferentes características dos pacientes.
Os resultados de eficácia deste estudo são sumarizados na tabela a seguir:
| Filgrastim (substância ativa) (n=160) | Comparador * (n=80) | |
| DSN (dias) | ||
| Ciclo 1 | 0,5 | 0,3 |
| ? DSN, dias (95% CI)* | 0,157 (-0,114, 0,428) | |
| Ciclo 4 | 0,4 | 0,3** |
| ANC nadir, 109/L | ||
| Ciclo 1 | 2,1 | 2,9 |
| Ciclo 4 | 2,3 | 3,2** |
| Tempo de recuperação ANC (dias) | ||
| Ciclo 1 | 6,3 | 4,5 |
| Ciclo 4 | 6,4 | 4,5** |
| Incidência de neutropenia febril‡ (%) | ||
| Ciclo 1 | 15,0 | 8,8 |
| Ciclo 4 | 4,3 | 3,3** |
| Todos os ciclos | 33,1 | 23,8 |
† Estimativa de análise de covariância (ANCOVA) e o intervalo de confiança de 95% para a diferença entre o Filgrastim (substância ativa) e o medicamento comparador no ciclo 1;
*Pacientes deste grupo receberam o medicamento comparador no ciclo 1 e o Filgrastim (substância ativa) nos ciclos seguintes;
**Após migrar do medicamento comparador para o Filgrastim (substância ativa);
‡ Observado ou definido em protocolo;
DSN: Duração de neutropenia severa;
ANC: Contagem absoluta de neutrófilos;
FN: Incidência de neutropenia febril.
Linfomas Não-Hodgkin
Estudo clínico fase III, multinacional, multicêntrico e randomizado, de comparação entre Filgrastim (substância ativa) e medicamento comparador, conduzido em pacientes com linfoma não-Hodgkin. Durante o estudo, os pacientes estavam sob tratamento quimioterápico, de no máximo 6 ciclos, com ciclofosfamida, doxorrubicina, vincristina e prednisona (CHOP). Tratamento adicional com rituximabe (anticorpo monoclonal anti-CD20) foi utilizado a critério de médico.
Os resultados de eficácia basearam-se nas seguintes determinações:
Duração de neutropenia severa (DSN):
A duração de neutropenia severa no ciclo 1 foi de 0,5 dias para pacientes tratados com o Filgrastim (substância ativa) e 0,9 dias para os pacientes tratados com o medicamento comparador. A estimativa de análise de covariância (ANCOVA) entre o Filgrastim (substância ativa) e o medicamento comparador foi de -0,378 dias. O intervalo de confiança de 95% (-0,837 a 0,081 dias) foi incluído na faixa pré-definida de equivalência (-1 a 1 dia), indicando que a duração de neutropenia severa (DSN) foi similar para o Filgrastim (substância ativa) e o medicamento comparador (p=0.11). A duração de neutropenia severa (DSN) no ciclo 4, após os pacientes migrarem do Filgrastim (substância ativa) para o medicamento comparador foi de, respectivamente, 0,2 e 0,7 dias para o grupo que recebeu Filgrastim (substância ativa) e o grupo que recebeu o medicamento comparador.
Contagem absoluta de neutrófilos (ANC):
O perfil de contagem absoluta de neutrófilos foi similar em todos os ciclos para o medicamento comparador e o Filgrastim (substância ativa). No ciclo 1, houve um aumento inicial de ANC significativo atingindo contagem máxima no 4° dia e subsequente diminuição no 9° dia. O ANC atingiu uma segunda contagem máxima no 11° dia e retornou próxima a contagem basal, gradualmente, até o 21° dia. A contagem absoluta de neutrófilos foi comparável entre os grupos que receberam o medicamento comparador e o Filgrastim (substância ativa) no ciclo 1 (1,7 vs. 1,1 x 109/L) e após migrarem do Filgrastim (substância ativa) para o medicamento comparador no ciclo 4 (2,1 vs. 1,8 x 109/L). O tempo médio de recuperação na contagem absoluta de neutrófilos nos pacientes que receberam o Filgrastim (substância ativa) e o medicamento comparador foi respectivamente de 6,0 e 6,7 dias no ciclo 1, e 4,9 e 6,1 dias no ciclo 4.
Incidência de neutropenia febril (FN):
No ciclo 1, a incidência observada ou definida em protocolo foi de 11,1% no grupo que recebeu o Filgrastim (substância ativa) e 20,7% no grupo que recebeu o medicamento comparador (p=0.12). As taxas de incidência no ciclo 4 foram respectivamente 31,7% e 41,4% (p=0.21).
Este estudo clínico fase III foi conduzido em pacientes com linfoma não-Hodgkin tratados com o regime quimioterápico CHOP, com ou sem rituximabe. Os resultados do estudo confirmaram que a profilaxia primária com o Filgrastim (substância ativa) é tão eficaz quanto com o medicamento comparador na redução da duração de neutropenia severa e na incidência de neutropenia febril. O perfil de contagem absoluta de neutrófilos foi similar entre ambos os medicamentos no ciclo 1.
Os resultados de eficácia resumidos estão demonstrados a seguir:?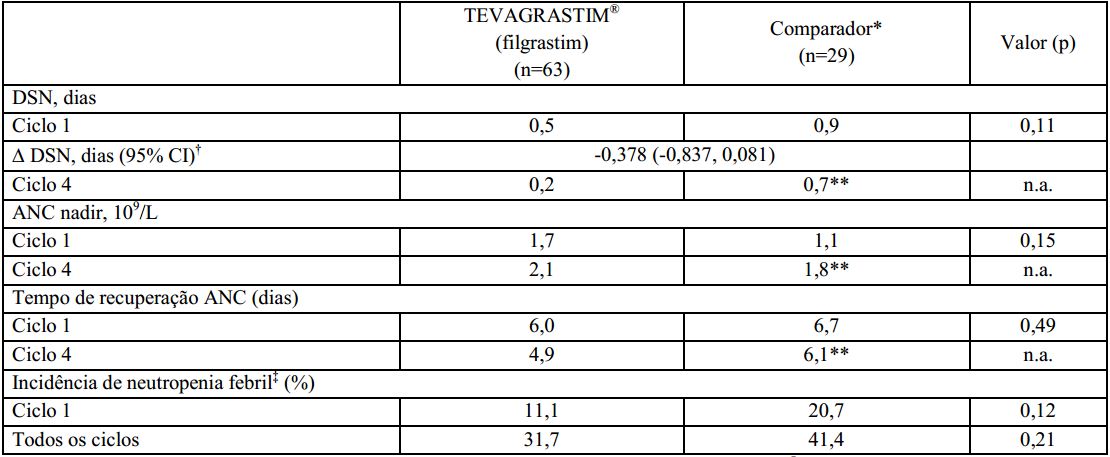 *Pacientes deste grupo receberam o medicamento comparador no ciclo 1 e o Filgrastim (substância ativa) nos ciclos seguintes;
*Pacientes deste grupo receberam o medicamento comparador no ciclo 1 e o Filgrastim (substância ativa) nos ciclos seguintes;
† Estimativa de análise de covariância (ANCOVA) e o intervalo de confiança de 95% para a diferença entre o Filgrastim (substância ativa) e o medicamento comparador no ciclo 1;
**Após migrar do medicamento comparador para o Filgrastim (substância ativa);
‡ Observado ou definido em protocolo;
DSN: Duração de neutropenia severa;
ANC: Contagem absoluta de neutrófilos;
FN: Incidência de neutropenia febril;
n.a.: Não avaliado
Característias Farmacológicas
Grupo Farmacoterapêutico:
Imunoestimuladores, Fatores Estimuladores de Colônias.
Código ATC: L03AA02.
Propriedades farmacodinâmicas
Filgrastim (substância ativa), é uma glicoproteína que regula a produção e a liberação de neutrófilos funcionais da medula óssea. O Filgrastim (substância ativa) provoca, em um período de 24 horas, um aumento significativo da contagem de neutrófilos no sangue periférico, com pequenos aumentos de monócitos. Em alguns pacientes com neutropenia crônica grave, o Filgrastim (substância ativa) pode também induzir um pequeno aumento do número de eosinófilos e basófilos circulantes em relação aos valores basais; alguns destes pacientes podem apresentar eosinofilia ou basofilia antes do tratamento. Elevações nas contagens de neutrófilos são dosedependentes nas doses recomendadas. Os neutrófilos produzidos em resposta ao Filgrastim (substância ativa) apresentam função normal ou aumentada, conforme demonstrado em testes de funções fagocítica e quimiostática. Após o término do tratamento com Filgrastim (substância ativa), a contagem de neutrófilos circulantes diminui em 50% dentro de 1 a 2 dias, e para níveis normais dentro de 1 a 7 dias.
O uso de Filgrastim (substância ativa) em pacientes submetidos à quimioterapia citotóxica leva a reduções significativas na incidência, gravidade e duração da neutropenia e da neutropenia febril. O tratamento com Filgrastim (substância ativa) reduz significativamente a duração da neutropenia febril, a utilização de antibióticos e o tempo de hospitalização após quimioterapia de indução para leucemia mieloide aguda ou terapia mieloablativa seguida de transplante de medula óssea. A incidência de relatos de febre e infecções não foi reduzida em nenhum destes quadros clínicos. A duração da febre não diminuiu nos pacientes que receberam terapia mieloablativa seguida de transplante de medula óssea.
A utilização de Filgrastim (substância ativa), isoladamente ou após quimioterapia, mobiliza as células progenitoras hematopoiéticas para o sangue periférico. Estas células progenitoras do sangue periférico (CPSP) autólogas podem ser coletadas e infundidas após terapia citotóxica de dose elevada, em substituição ou em adição ao transplante de medula óssea. A infusão de CPSP acelera a recuperação hematopoiética, reduzindo a duração do risco de complicações hemorrágicas e a necessidade de transfusões de plaquetas.
Os receptores de CPSP alogênicas mobilizadas com Filgrastim (substância ativa) tiveram uma recuperação hematológica significativamente mais rápida, levando a uma diminuição significativa do tempo de recuperação de plaquetas quando comparado com o transplante alogênico de medula óssea.
Um estudo Europeu retrospectivo que avaliou o uso de fator estimulador de colônias de granulócitos (G-CSF) após transplante alogênico de medula óssea em pacientes com leucemia aguda sugeriu risco aumentado de doença do enxerto contra hospedeiro (graft-versus-host disease - GVHD), mortalidade relacionada ao tratamento (TRM) e mortalidade quando o G-CSF foi administrado. Outro estudo retrospectivo internacional, conduzido em pacientes com leucemias mieloides aguda e crônica, não foi observado risco de GVHD, TRM e mortalidade. Uma metanálise de estudos de transplantes alogênicos, incluindo resultados de nove estudos randomizados prospectivos, 8 estudos retrospectivos e 1 estudo de caso-controle, não detectou efeito sobre o risco de GVHD aguda, GVHD crônica ou mortalidade relacionada ao tratamento.
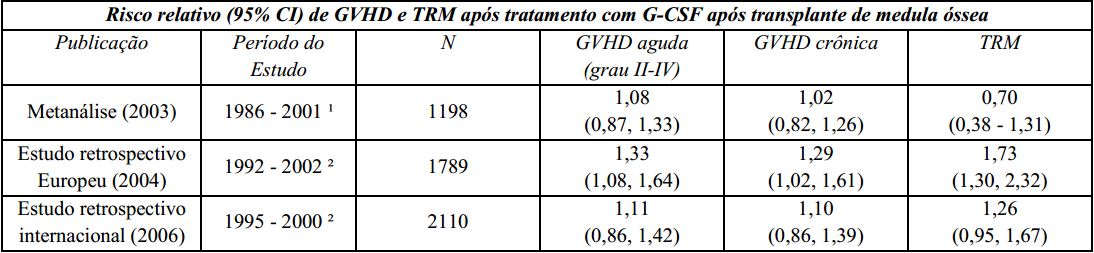 GVHD: doença do enxerto contra hospedeiro (graft-versus-host disease);
GVHD: doença do enxerto contra hospedeiro (graft-versus-host disease);
TRM: mortalidade relacionada ao tratamento;
G-CSF: fator estimulador de colônias de granulócitos;
¹ A análise incluiu estudos envolvendo transplante de medula óssea durante este período; alguns estudos utilizaram GM-CSF (fator estimulador de colônias de macrófagos e granulócitos);
² A análise incluiu pacientes recebendo transplante de medula óssea durante este período.
Previamente ao transplante de CPSP alogênicas, a utilização de Filgrastim (substância ativa) para a mobilização de CPSP em doadores saudáveis permite um cultivo de 4 x 106 células CD34+ /kg de peso corporal do receptor na maioria dos doadores, após duas leucaféreses. Para estes doadores saudáveis é dada uma dose de 10 mcg/kg/dia, administrada por via subcutânea durante 4 a 5 dias consecutivos.
O uso de Filgrastim (substância ativa) em pacientes com neutropenia crônica grave (neutropenia congênita grave, neutropenia cíclica e neutropenia idiopática), induz um aumento sustentado das contagens absolutas de neutrófilos no sangue periférico e uma redução das infecções e eventos relacionados.
O Filgrastim (substância ativa), assim como com outros fatores de crescimento hematopoiéticos, demonstrou in vitro propriedades estimuladoras sobre as células endoteliais humanas.
Propriedades Farmacocinéticas
A depuração de Filgrastim (substância ativa), tanto após administração subcutânea como intravenosa, demonstrou seguir uma farmacocinética de primeira ordem. A meia-vida de eliminação sérica do Filgrastim (substância ativa) é de aproximadamente 3,5 horas, com uma taxa de depuração de aproximadamente 0,6mL/min/kg. A infusão contínua com Filgrastim (substância ativa) durante um período de até 28 dias, em pacientes em recuperação de transplante autólogo de medula óssea, não apresentou evidência de acumulação do fármaco e de meias-vidas comparáveis. Há uma correlação linear positiva entre a dose e a concentração sérica de Filgrastim (substância ativa), se administrado por via intravenosa ou por via subcutânea. Após administração subcutânea das doses recomendadas, as concentrações séricas mantiveramse acima dos 10 ng/mL, durante 8 a 16 horas. O volume de distribuição no sangue é aproximadamente de 150mL/kg.
Em pacientes com câncer, o perfil farmacocinético do Filgrastim (substância ativa) e do medicamento comparador foi comparável após administração única e após administrações múltiplas por via subcutânea.
Dados de Segurança Pré-Clínicos
Os dados pré-clínicos não revelaram risco especial para humanos com base em estudos convencionais de segurança farmacológica, genotoxicidade e tolerância local.
Os dados pré-clínicos de estudos convencionais de toxicidade de doses múltiplas demonstraram os efeitos farmacológicos esperados, incluindo aumento da contagem leucocitária, hiperplasia mieloide da medula óssea, hematopoiese extramedular e dilatação esplênica.
Não foram observados efeitos sobre a fertilidade de ratos do sexo masculino e feminino, assim como sobre a gestação em ratos. Não existe evidência a partir dos estudos conduzidos em ratos e coelhos de que o Filgrastim (substância ativa) seja teratogênico. Foi observada incidência aumentada de perda embrionária em coelhos, porém não foi observada má formação.
Cuidados de Armazenamento
Granulokine deve ser armazenado sob refrigeração, entre (2°C e 8°C).
A exposição acidental a temperaturas congelantes não afeta desfavoravelmente a estabilidade do produto.
O profissional da saúde saberá como armazenar o medicamento depois de aberto.
Número de lote e datas de fabricação e validade: Vide embalagem.
Após preparo, manter sob refrigeração de (2°C a 8°C) por até 24 horas.
Soluções diluídas de Granulokine não devem ser preparadas mais de 24 horas antes da administração.
Para sua segurança, mantenha o medicamento na embalagem original.
Característica física
A solução de Granulokine é límpida e incolor.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartadosbno esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS-1.0244.0006
Farm. Resp.:
Monica Carolina Dantas Pedrazzi CRF-SP 30.103
Importado por:
Amgen Biotecnologia do Brasil Ltda.
Rua Patrícia Lúcia de Souza, 146
Taboão da Serra – SP
CNPJ: 18.774.815/0001-93
Fabricado por:
F. Hoffmann-La Roche Ltd, Kaiseraugst, Suíça
Embalado por:
Amgen Europe B.V., Breda, Holanda
Sac: 0800 264 0800
Uso restrito a hospitais.
Venda sob prescrição médica.