Prograf Cápsula 1mg, caixa com 100 cápsulas duras na Pharmed
Prograf - 1Mg Com 100 Cápsulas

Prograf Cápsula 1mg, caixa com 100 cápsulas duras na Pharmed
Não encontramos este produto para a Pharmed no Cliquefarma.
Confira os menores preços para Prograf - 1Mg Com 100 Cápsulas
Prograf - 1Mg Com 100 Cápsulas
66.33%
R$ 547,79
OncoExpresso Medicamentos
66.26%
R$ 549,00
Sampharma Medicamentos Especiais
65.89%
R$ 554,90
Maranata Medicamentos
60.49%
R$ 642,90
Farma Visa
60.25%
R$ 646,70
Para que serve
Prograf é recomendado depois que você é submetido a um transplante de rim ou fígado, a fim de evitar que o seu organismo rejeite o órgão transplantado. Recomenda-se que Prograf seja utilizado concomitantemente com corticosteroides.
Como Prograf funciona?
Prograf é um medicamento que reduz a resposta do seu sistema imunológico e atua como medicamento anti-rejeição, evitando que o seu organismo rejeite o órgão que você recebeu.
Contraindicação
Cápsula
Este medicamento é contraindicado para pacientes com hipersensibilidade (alergia) ao tacrolimo ou a qualquer componente da fórmula do medicamento.
Solução injetável
Você não deve tomar Prograf se for alérgico ao tacrolimo (ingrediente ativo) ou a qualquer componente da fórmula do medicamento, em particular ao óleo de rícino hidrogenado polioxietileno.
Como usar
Prograf pode ser administrado por via intravenosa ou oral. De forma geral, a administração pode iniciar-se por via oral; se necessário, através da administração do conteúdo da cápsula em suspensão em água, por entubação nasogástrica.
Tacrolimo não é compatível com o plástico PVC. Tubos, seringas e outros equipamentos usados para preparar ou administrar a suspensão do conteúdo das cápsulas de Prograf, não devem conter PVC em sua composição.
Cápsula
Sempre tome Prograf exatamente como o seu médico lhe orientou. Você deve verificar com o seu médico ou farmacêutico caso não tenha certeza de alguma orientação recebida. Este medicamento só deve ser prescrito por um médico com experiência no tratamento de pacientes transplantados.
As cápsulas de Prograf devem ser tomadas duas vezes ao dia (por exemplo, de manhã e à noite), com intervalos de 12 horas, por via oral.
As cápsulas de Prograf devem ser tomadas imediatamente após a remoção do blister. As cápsulas devem ser ingeridas com líquido (de preferência água) e com o estômago vazio ou, pelo menos, 1 hora antes das refeições ou de 2 a 3 horas após as refeições, para conseguir uma máxima absorção do medicamento.
Este medicamento não deve ser partido, aberto ou mastigado.
Solução injetável
A preparação e aplicação do medicamento devem ser feitas exclusivamente por profissional de saúde com experiência e capacitação.
Prograf solução injetável é somente para infusão intravenosa e deve ser reservado para pacientes impossibilitados de tomar Prograf cápsulas por via oral.
Se a terapia intravenosa for necessária, recomenda-se conversão da terapia intravenosa com Prograf solução injetável para a terapia oral com Prograf cápsulas tão logo o paciente consiga tolerar a administração oral. Isso geralmente ocorre dentro de 2-3 dias.
A terapia intravenosa não deve prosseguir por mais de 7 dias.
Em pacientes que receberam uma infusão intravenosa, a primeira dose oral do tratamento deve ser administrada de 8 - 12 horas após a interrupção da infusão intravenosa.
Caso a solução diluída de Prograf seja administrada acidentalmente através uma artéria ou por via perivasal, pode ocorre irritação no local da aplicação.
Posologia
Cápsula
A dosagem inicial é estabelecida pelo médico, de acordo com seu peso e com o órgão que você recebeu.
Resumo das recomendações de dose oral inicial e as concentrações no sangue total
|
População de pacientes | Dose oral inicial* |
Concentrações mínimas no sangue total |
|
Adultos - Transplante renal | 0,2 mg/kg/dia |
Mês 1– 3 : 7 – 20 ng/mL |
|
Mês 4 – 12 : 5 – 15 ng/mL | ||
|
Adultos - Transplante hepático | 0,10 – 0,15 mg/kg/dia |
Mês 1–12 : 5 – 20 ng/mL |
|
Crianças - Transplante hepático | 0,15 – 0,20 mg/kg/dia |
Mês 1– 12 : 5 – 20 ng/mL |
*Dividida em duas doses, administradas a cada 12 horas.
Transplante hepático
Se possível, o tratamento será iniciado com Prograf cápsulas. Caso a terapia intravenosa seja necessária, a mudança de Prograf solução injetável para cápsulas é recomendada assim que a terapia oral puder ser tolerada. Isso usualmente ocorre em 2-3 dias. A dose inicial de Prograf não deve ser administrada antes de 6 horas depois do transplante. Se você estiver recebendo infusão intravenosa, a primeira dose da terapia oral deve ser administrada de 8 – 12 horas depois da descontinuação da infusão intravenosa.
A dose oral inicial de Prograf cápsulas é de 0,10 – 0,15 mg/dia, dividida em duas doses, com intervalo de 12 horas.
Em pacientes receptores de transplante hepático, a administração concomitante com suco de toranja (grapefruit) aumenta as concentrações mínimas de tacrolimo no sangue. Seu médico irá ajustar a dose com base na avaliação clínica de rejeição e tolerabilidade. Doses menores de Prograf podem ser suficientes como terapia de manutenção. Uma terapia conjunta com corticosteroides adrenais é recomendada logo após o transplante.
Transplante de Rim
No transplante de rim, a dose oral inicial recomendada de Prograf cápsulas é de 0,2 mg/kg/dia administrada a cada 12 horas, em duas doses. A dose inicial de Prograf pode ser administrada 24 horas após o transplante, mas deve ser adiada até a recuperação da função renal (conforme indicado, por exemplo, por creatinina sérica ? 4 mg/dL). Pacientes negros podem precisar de doses mais elevadas para alcançar concentrações sanguíneas comparáveis.
Pacientes pediátricos
Crianças que receberam um transplante de fígado e que não apresentavam função renal ou hepática prejudicada antes da cirurgia precisam e toleram receber doses mais elevadas do que adultos para atingirem concentrações semelhantes no sangue. Portanto, recomenda-se que a terapia seja iniciada em crianças com uma dose intravenosa inicial de 0,03 – 0,05 mg/kg/dia ou uma dose oral inicial de 0,15 – 0,20 mg/kg/dia.
Ajustes na dose podem ser necessários. A experiência em pacientes pediátricos receptores de transplante de rim é limitada.
Conversão de um tratamento imunossupressor para outro
Prograf não deve ser usado simultaneamente com ciclosporina.
Prograf ou ciclosporina devem ser descontinuados no mínimo 24 horas antes de iniciar o outro medicamento. Na presença de concentrações elevadas de Prograf ou ciclosporina, a administração do medicamento deve, em geral, ser adiada.
Solução injetável
A dose inicial de Prograf não deve ser administrada antes de 6 horas depois do transplante.
A dose inicial de Prograf solução injetável é 0,03 - 0,05 mg/kg/dia, administrada sob a forma de infusão intravenosa contínua, tanto para transplante hepático quanto para transplante renal. Os pacientes adultos devem receber os limites inferiores da faixa de dose.
Terapia concomitante com corticosteroides adrenais é recomendada logo após o transplante.
Pacientes Pediátricos
Pacientes pediátricos receptores de transplante hepático sem disfunção renal ou hepática preexistente requereram e toleraram doses mais elevadas que os adultos para alcançar concentrações sanguíneas similares. Portanto, é recomendado que a terapia seja iniciada em pacientes pediátricos com uma dose intravenosa inicial de 0,03 - 0,05 mg/kg/dia. Ajustes na dose podem ser necessários.
A experiência em pacientes pediátricos receptores de transplante de rim é limitada.
Pacientes geriátricos
Não há evidências atualmente disponíveis que a dose de Prograf deva ser ajustada em pacientes geriátricos.
Pacientes com disfunção renal ou hepática
Devido ao potencial de toxicidade aos rins, pacientes com função renal ou hepática prejudicada devem receber doses mais baixas no intervalo de dose intravenosa e oral recomendado. Reduções adicionais na dose abaixo dessas faixas podem ser necessárias. Em geral, a terapia com Prograf deve ser adiada por até 48 horas ou mais se você apresentar redução na frequência de micção após a cirurgia (oligúria pós-operatória).
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Prograf?
Se você se esquecer de tomar Prograf, tome a dose recomendada assim que lembrar. Se estiver muito próximo ao horário da dose seguinte, pule a dose que esqueceu de tomar e tome a dose seguinte no horário recomendado. Não tome uma dose dobrada para compensar a dose esquecida.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Gerenciamento da imunossupressão (redução da atividade ou da eficiência do sistema imune)
Somente médicos com experiência em terapia imunossupressora e tratamento de pacientes com transplante de órgãos devem prescrever Prograf. Pacientes que usam o medicamento devem ser monitorados em instituições com recursos médicos e laboratoriais adequados. O médico responsável pela terapia de manutenção deve ter todas as informações necessárias para monitorar o paciente.
Monitoramento de rotina
Você deve manter contato regular com seu médico. Ocasionalmente, seu médico pode precisar de exames de sangue, urina, do coração ou dos olhos, para definir a dose certa de Prograf.
Informe o seu médico se alguma das seguintes situações se aplicarem a você
- Se você tem ou já teve problemas de fígado;
- Se tiver diarreia por mais de um dia;
- Se você se sentir forte dor abdominal acompanhada ou não de outros sintomas, tais como calafrios, febre, náuseas ou vômitos;
- Se você tem uma alteração de atividade elétrica do coração chamado de "prolongamento do intervalo QT”.
Uma vez que Prograf pode causar alterações no funcionamento do rim ou do fígado, seu médico solicitará exames de sangue com frequência.
Durante o período inicial após o transplante, os seguintes parâmetros devem ser monitorados de forma rotineira pelo seu médico
- Pressão arterial para possível hipertensão (aumento da pressão sanguínea);
- Exames do coração, como eletrocardiograma (ECG);
- Estado neurológico e visual;
- Níveis de glicose no sangue (glicemia em jejum), para investigação de possível aumento de glicose sanguínea (hiperglicemia) ou de diabetes mellitus;
- Níveis de eletrólitos no sangue (particularmente do potássio, para investigação de possível aumento dos níveis de potássio no sangue - hiperpotassemia);
- Testes de função do fígado e dos rins;
- Parâmetros hematológicos;
- Valores de coagulação e proteínas presentes no plasma do sangue.
Se alterações clinicamente relevantes forem observadas, deve-se considerar o ajuste do esquema imunossupressor.
Os níveis de tacrolimo no sangue podem variar significativamente durante episódios de diarreia. Por essa razão, recomenda-se monitoramento extra da concentração de tacrolimo no sangue durante esses episódios.
Erros de Medicações
Erros de medicação, incluindo a substituição inadvertida, não intencional ou não supervisionada de formulações de tacrolimo com liberação imediata ou prolongada foram observados. Isso resultou em reações adversas graves, incluindo rejeição do órgão transplantado, ou outras reações adversas que poderiam ocorrer como consequência da exposição insuficiente ou excessiva ao tacrolimo. Os pacientes devem ser mantidos sob uma única formulação de tacrolimo com o regime de dose diário correspondente. Alterações na formulação ou no regime de dose devem ser feitas somente sob a supervisão atenta de um médico especialista em transplante.
Hipertrofia do Miocárdio (aumento do coração)
Hipertrofia ventricular ou hipertrofia do septo, tipos de doenças cardíacas caracterizadas pelo aumento do coração, têm sido observadas em raras ocasiões com o uso de tacrolimo. A maioria dos casos foi reversível, ocorrendo principalmente em crianças com concentrações sanguíneas mínimas de tacrolimo muito superiores aos níveis máximos recomendados.
Portanto, os pacientes de alto risco, principalmente crianças que receberam imunossupressão substancial, devem ser monitorados utilizando procedimentos como ecocardiografia ou eletrocardiograma (ECG) pré e pós-transplante (por exemplo, inicialmente aos três meses e, então, aos 9-12 meses).
Se você tiver aumento do coração durante o tratamento com Prograf, seu médico poderá reduzir a dose ou interromper o tratamento.
Hipertensão (aumento da pressão sanguínea)
Aumento da pressão sanguínea (hipertensão arterial) é um efeito adverso comum inerente à terapia com Prograf e você pode necessitar de tratamento anti-hipertensivo.
O controle da pressão sanguínea pode ser obtido com o uso de qualquer agente antihipertensivo comum, embora se deva ter cautela ao usar determinados agentes anti-hipertensivos associados ao aumento dos níveis de potássio no sangue.
Caso você apresente aumento da pressão sanguínea durante o tratamento com Prograf, seu médico poderá receitar medicamentos anti-hipertensivos.
Prolongamento do intervalo QT (alterações na atividade elétrica do coração)
Prograf pode causar arritmias (irregularidades nas batidas do coração), como alterações no exame de eletrocardiograma (alterações na atividade elétrica do coração, como prolongamento do intervalo QT e torsade de pointes). Assim, o seu médico irá lhe informar sobre as precauções a serem tomadas caso você utilize Prograf e tenha tais situações clínicas.
Em pacientes com insuficiência cardíaca congestiva, diminuição da frequência cardíaca (bradiarritmia), aqueles que tomam certos medicamentos que alteram a atividade elétrica do coração, e aqueles com diminuição dos níveis sanguíneos de potássio, cálcio ou magnésio, deve-se considerar a realização de eletrocardiograma e monitoramento de eletrólitos (magnésio, potássio, cálcio) periodicamente durante o tratamento com Prograf.
Infecções Graves
O tratamento com Prograf diminuirá a sua imunidade e você estará mais sujeito a contrair infecções por bactérias, vírus, fungos ou protozoários. Por isso, é importante avisar seu médico se você tiver febre.
Este medicamento pode aumentar o risco de infecções. Informe ao seu médico qualquer alteração no seu estado de saúde.
Medicamentos imunossupressores, como Prograf, podem ativar focos primários de tuberculose.
Os médicos que monitoram pacientes imunossuprimidos devem estar alertas quanto à possibilidade de surgimento de doença ativa e devem, portanto, tomar todas as precauções cabíveis para o diagnóstico e o tratamento precoce. Este medicamento pode aumentar o risco de infecção. Informe seu médico sobre qualquer mudança na sua condição de saúde.
Este medicamento pode aumentar o risco de sangramento em caso de dengue.
Diabetes mellitus pós-transplante (DMPT)
Nos pacientes submetidos a transplante de rim e fígado, o tratamento com Prograf pode provocar o aparecimento de diabetes, que se manifesta por um aumento da frequência de micção, aumento da sede ou do apetite. Portanto, avise seu médico se apresentar algum desses sintomas.
Nefrotoxicidade (toxicidade dos rins)
Deve-se tomar cuidado ao utilizar tacrolimo com outros medicamentos nefrotóxicos (que causam danos aos rins). Em especial, para evitar nefrotoxicidade excessiva, Prograf não deve ser usado simultaneamente com a ciclosporina ou outros medicamentos com efeitos nefrotóxicos conhecidos.
O uso de tacrolimo pode resultar em comprometimento da função renal em pacientes transplantados. O comprometimento renal é geralmente reversível, podendo resultar no aumento dos níveis sanguíneos de creatinina, potássio e ácido úrico e na diminuição da secreção de ureia.
Pacientes com comprometimento renal devem ser monitorados atentamente, já que pode ser necessária a redução da dose de tacrolimo ou a suspensão temporária do tratamento.
Neurotoxicidade (toxicidade do sistema nervoso)
O tacrolimo pode causar neurotoxicidade (alteração da atividade normal do sistema nervoso, causando dano ao tecido nervoso) especialmente quando usado em doses elevadas.
A maioria das neurotoxicidades graves incluem encefalopatia posterior reversível (PRES) doenças que afetam o sistema nervoso, por exemplo, delírio e coma.
Caso os pacientes que estejam tomando tacrolimo apresentem sintomas indicativos de PRES, tais como dor de cabeça, alteração do estado mental, convulsões e distúrbios visuais, um exame radiológico (por exemplo, ressonância magnética) deve ser realizado. Caso seja diagnosticado PRES, aconselha-se o controle adequado da pressão arterial e a suspensão imediata do tacrolimo. A maioria dos pacientes recupera-se completamente após serem tomadas medidas adequadas.
Convulsões ocorreram em pacientes adultos e pediátricos que receberam Prograf.
Neurotoxicidades menos graves, por exemplo, tremores, parestesias, dor de cabeça e outras alterações na função motora, estado mental e função sensorial. Tremor e dor de cabeça foram associados à altas concentrações de tacrolimo no sangue e podem responder ao ajuste de dose.
Se você apresentar tremores, dor de cabeça e alteração dos movimentos durante o tratamento com Prograf, informe seu médico, pois poderá ser necessário ajustar a dose que você está tomando.
Hiperpotassemia (aumento do nível de potássio na corrente sanguínea)
Hiperpotassemia (aumento do nível de potássio na corrente sanguínea) foi relatada com o uso de tacrolimo. Assim, o nível de potássio no sangue deve ser monitorado. O uso de diuréticos poupadores de potássio ou o alto consumo de potássio deve ser evitado durante o tratamento com Prograf.
Distúrbios linfoproliferativos (alguns tipos de câncer do sistema linfático) e outras malignidades
Como resultado da imunossupressão, a suscetibilidade à infecções e possível desenvolvimento de linfoma e outras neoplasias malignas (alguns tipos de câncer) em indivíduos tratados com tacrolimo pode ser maior que na população sadia normal.
Um distúrbio linfoproliferativo relacionado à infecção por um vírus chamado Epstein-Barr (EBV) foi relatado em receptores de órgãos transplantados imunossuprimidos. O risco de distúrbios linfoproliferativos é maior em crianças menores que estão sob o risco da infecção primária por EBV enquanto estão imunossuprimidas ou que passam a receber Prograf após um longo período de terapia de imunossupressão. Foi relatado que a redução ou descontinuação da imunossupressão pode levar à regressão das lesões.
O aumento da incidência de neoplasias malignas (alguns tipos de câncer) é uma complicação reconhecida da imunossupressão em receptores de transplante de órgãos. As formas mais comuns de neoplasia são linfomas não-Hodgkin e doenças malignas de pele. Portanto, a exposição ao sol e à luz ultravioleta deve ser limitada através do uso de roupas protetoras e um protetor solar com alto fator de proteção.
Imunizações
Algumas vacinas são contraindicadas para quem está tomando medicamentos imunossupressores. Antes de tomar qualquer vacina, informe ao profissional de saúde que você está tomando medicamento imunossupressor.
Durante o tratamento com Prograf, você não deve tomar nenhuma vacina sem antes consultar um médico, pois a vacina pode não funcionar como deveria ou pode provocar efeitos colaterais.
Aplasia pura da série vermelha (interrupção na produção de células vermelhas do sangue)
Casos de um tipo de anemia conhecida como aplasia pura da série vermelha (PCRA) foram relatados em pacientes tratados com tacrolimo. Todos os pacientes apresentavam fatores de risco para PRCA, tais como infecção por parvovírus B19, doença de base ou medicamentos concomitantes associados com PRCA.
Se a PRCA for diagnosticada, deve-se considerar a interrupção do tratamento com Prograf.
Perfuração gastrointestinal
Casos de perfuração gastrointestinal foram relatados em pacientes tratados com tacrolimo.
Uma vez que a perfuração gastrointestinal é um evento clinicamente importante que pode resultar em doença grave ou ameaça à vida, se você apresentar qualquer sintoma contate seu médico imediatamente para que tratamentos adequados, incluindo cirurgia, sejam considerados.
Excipientes
Cápsula
Prograf de 1 mg
Este produto contém 61,35 mg de lactose monoidratada por cápsula; portanto, deve ser usado com cautela por pessoas com diabetes.
Prograf de 5 mg
Este produto contém 123,60 mg de lactose monoidratada por cápsula; portanto, deve ser usado com cautela por pessoas com diabetes.
Solução injetável
Prograf solução injetável contém álcool.
O teor de álcool presente na solução injetável de Prograf (etanol - 638 mg por mL) deve ser levado em consideração.
Prograf solução injetável contém óleo de rícino hidrogenado polioxietileno, cujos relatos apontam como a causa de reações anafiláticas. Dessa forma, deve-se ter cautela com aqueles pacientes que receberam previamente preparações contendo derivados de óleo de rícino hidrogenado por infusão e àqueles pacientes com predisposição alérgica a esse composto.
O risco de anafilaxia pode ser reduzido através da infusão lenta de PROGRAF solução injetável ou através da administração prévia de um anti-histamínico. O paciente deve ser observado durante os primeiros 30 minutos da infusão para possíveis reações anafiláticas.
Conversão de ciclosporina para tacrolimo
Tacrolimo não deve ser usado simultaneamente com ciclosporina. O uso de Prograf ou ciclosporina deve ser interrompido pelo menos 24 horas antes do início do uso do outro medicamento. Em situações de concentrações elevadas de Prograf ou de ciclosporina, o uso do outro medicamento deve ser adiado.
A administração concomitante de ciclosporina e tacrolimo não é recomendada.
Efeito sobre a capacidade de dirigir e operar máquinas
O tacrolimo pode causar distúrbios visuais e neurológicos. Esse efeito pode ser acentuado se o tacrolimo for administrado em associação com álcool.
Pacientes com função renal e hepática prejudicada
Se você recebeu um transplante de fígado e não está bem, o uso de Prograf pode estar associado a um maior risco para desenvolvimento de insuficiência renal relacionada aos níveis elevados de tacrolimo no sangue total. Nesse caso, o médico fará o monitoramento atento da situação até o final do tratamento e, se necessário, fará ajustes à dose de Prograf.
Pacientes pediátricos
Pacientes pediátricos, geralmente, requerem doses maiores de Prograf para manter concentrações sanguíneas similares as de adultos.
Raça
A comparação retrospectiva dos pacientes negros e caucasianos submetidos ao transplante de rim indicou que os pacientes negros necessitavam de doses mais elevadas de tacrolimo para atingirem concentrações mínimas semelhantes.
Pacientes idosos
Não foram observadas diferenças gerais na segurança ou eficácia entre pacientes idosos e mais jovens, mas a sensibilidade maior de alguns pacientes mais idosos não pode ser descartada. Em geral, recomenda-se cautela quanto à seleção de dose para pacientes geriátricos, refletindo a maior frequência de redução nas funções hepática e renal e de doenças concomitantes ou uso de outros medicamentos.
Gravidez
Não existem estudos adequados e bem controlados em mulheres grávidas. O tacrolimo é transferido através da placenta. Prograf deverá ser usado durante a gravidez somente se o benefício para a mãe justificar o risco potencial ao feto.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação de um médico ou um cirurgião-dentista.
Lactação
Uma vez que o tacrolimo é excretado no leite humano, a amamentação deve ser interrompida durante o tratamento.
Durante o período de aleitamento materno ou doação de leite humano, só utilize medicamentos com o conhecimento do seu médico ou cirurgião-dentista, pois alguns medicamentos podem ser excretados no leite humano, causando reações indesejáveis no bebê.
Interações medicamentosas
Alguns medicamentos (inclusive fitoterápicos, como erva-de-são-joão e suplementos vitamínicos) e alguns alimentos podem interferir na ação de Prograf. Portanto, sempre verifique com seu médico se você pode tomar qualquer outro medicamento.
Os níveis de Prograf no sangue podem ser afetados por outros medicamentos que você tomar, e os níveis no sangue de outros medicamentos podem ser afetados pelo uso de Prograf, que pode precisar ser interrompido, ter a dose aumentada ou reduzida.
Em especial, você deve informar seu médico se estiver tomando ou tiver tomado recentemente medicamentos como
- Medicamentos usados para tratar problemas de pressão arterial ou de coração (diltiazem, nicardipina, nifedipina, verapamil);
- Antibióticos macrolídeos (claritromicina, eritromicina, troleandomicina, josamicina);
- Agentes antifúngicos (para tratar micoses) (clotrimazol, fluconazol, itraconazol, cetoconazol, voriconazol);
- Agentes gastrointestinais pró-cinéticos (cisaprida, metoclopramida);
- Inibidores de bomba de próton (lansoprazol, omeprazol);
- Outros medicamentos (amiodarona, bromocriptina, cloranfenicol, cimetidina, ciclosporina, danazol, etinilestradiol, prednisolona, metilprednisolona);
- Inibidores de protease do HIV (ritonavir, nelfinavir, saquinavir);
- Inibidores de protease do vírus da hepatite C (telaprevir, boceprevir), nefazodona, hidróxido de magnésio e alumínio, extrato de Schisandra sphenanthera;
- Medicamentos conhecidos como "estatinas" usados para o tratamento de colesterol e triglicerídeos.
Alguns medicamentos que podem diminuir a concentração de Prograf no sangue são
- Anticonvulsivantes (carbamazepina, fenobarbital, fenitoína);
- Antimicrobianos (rifabutina, caspofungina, rifampicina);
- Erva-de-são-joão;
- Sirolimo.
Informe seu médico se estiver tomando ou precisar tomar
- Ibuprofeno (usado para tratar febre, inflamação e dor);
- Anfotericina B (usado para tratar infecções fúngicas);
- Antivirais (usados para tratar infecções virais, por exemplo, aciclovir).
Eles podem agravar problemas renais ou do sistema nervoso quando tomados junto com Prograf.
O seu médico também necessita saber
- Se você está tomando suplementos de potássio ou certos diuréticos usados para a insuficiência cardíaca, hipertensão arterial e doença renal, (por exemplo, amilorida, triamtereno e espironolactona);
- Medicamentos anti-inflamatórios não esteroides (AINEs, por exemplo, ibuprofeno) usados para febre, inflamação e dor;
- Anticoagulantes (diluidores do sangue);
- Medicamentos orais para diabetes, enquanto você toma Prograf.
Durante o tratamento com Prograf, você não deve tomar nenhuma vacina sem antes consultar um médico, pois a vacina pode não funcionar como deveria.
Interação com alimentos
Foi relatado que a administração concomitante com suco de toranja (grapefruit) aumentou a concentração mínima de tacrolimo no sangue total em pacientes receptores de transplante hepático.
Portanto, você não deve tomar suco de toranja (grapefruit) durante o tratamento com Prograf.
A presença de alimento no estômago atrasa a absorção de tacrolimo.
Você não deve consumir bebidas alcoólicas durante o tratamento com Prograf.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como qualquer medicamento, Prograf pode provocar reações adversas, embora nem todas as pessoas os manifestem.
Prograf reduz o mecanismo de defesa (sistema imune) do seu corpo, que ficará prejudicado para o combate a infecções. Por isso, você pode estar mais propenso a infecções enquanto estiver tomando Prograf.
Podem ocorrer efeitos graves, incluindo reações alérgicas e anafiláticas. Foram relatados casos de tumores benignos e malignos após o tratamento com Prograf.
Foram relatados casos de aplasia eritrocítica pura (uma redução muito grave na contagem de glóbulos vermelhos), agranulocitose (uma contagem muito baixa de glóbulos brancos) e anemia hemolítica (número reduzido de glóbulos vermelhos devido a uma quebra anormal).
Reações adversas muito comuns (ocorrem em mais de 10% dos pacientes que utilizam este medicamento)
- Glicemia elevada;
- Diabetes mellitus insulino dependente e não insulino dependente;
- Nível elevado de potássio no sangue;
- Dificuldade para dormir;
- Tremor, dor de cabeça;
- Pressão arterial alta;
- Testes de função hepática anormais;
- Diarreia, náusea;
- Problemas renais.
Reações adversas comuns (ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento)
- Redução na contagem de glóbulos no sangue (plaquetas, glóbulos vermelhos ou brancos), elevação na contagem de glóbulos brancos, alterações na contagem de glóbulos vermelhos (observadas em exames de sangue);
- Nível reduzido de magnésio, fosfato, potássio, cálcio ou sódio no sangue, consumo excessivo de líquidos, elevação de ácido úrico ou lipídios no sangue, apetite reduzido, acidez elevada do sangue, outras alterações nos sais do sangue (observadas em exames de sangue);
- Sintomas de ansiedade, confusão e desorientação, depressão, alterações do humor, pesadelo, alucinação, distúrbios mentais;
- Ataques, distúrbios de consciência, formigamento e dormência (às vezes dolorosa) nas mão e nos pés, tontura, capacidade de escrita comprometida, distúrbios do sistema nervoso;
- Visão turva, sensibilidade elevada à luz, distúrbios nos olhos;
- Zumbido nos ouvidos;
- Fluxo sanguíneo reduzido nos vasos do coração, batidas mais rápidas do coração;
- Sangramento, bloqueio parcial ou completo dos vasos sanguíneos, pressão arterial baixa;
- Falta de ar, distúrbios dos tecidos respiratórios do pulmão, acúmulo de líquidos ao redor do pulmão, inflamação da faringe, tosse, sintomas semelhantes à gripe;
- Problemas estomacais, como inflamação ou úlcera causando dor abdominal ou diarreia, sangramento no estômago, inflamação ou úlcera na boca, acúmulo de líquidos no abdômen, vômito, dor abdominal, indigestão, prisão de ventre, flatulência, inchaço, fezes moles;
- Distúrbios no duto biliar, amarelamento da pele por problemas hepáticos, dano no tecido hepático e inflamação do fígado;
- Coceira, erupção cutânea, queda de cabelo, acne, sudorese elevada;
- Dor nas articulações, membros ou lombar, cãibras musculares e espasmos musculares;
- Função renal insuficiente, produção reduzida de urina, micção comprometida ou dolorosa;
- Fraqueza geral, febre, acúmulo de líquidos no corpo, dor e desconforto, aumento da enzima fosfatase alcalina no sangue, ganho de peso, sensação de temperatura desregulada;
- Função insuficiente do órgão transplantado;
- Percepção perturbada da temperatura corporal.
Reações adversas incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento)
- Alterações na coagulação do sangue, redução do número de todos os tipos de células sanguíneas (observadas em exames de sangue);
- Desidratação, incapacidade de urinar;
- Exames de sangue anormais: proteínas ou açúcar reduzidos, fosfato elevado, aumento da enzima lactato desidrogenase;
- Coma, sangramento no cérebro, acidente vascular cerebral, paralisia, distúrbio cerebral, anormalidades da fala e linguagem, problemas de memória;
- Turvamento do cristalino (olho), audição comprometida;
- Batimentos cardíacos irregulares, parada dos batimentos cardíacos, desempenho reduzido do coração, distúrbio do músculo cardíaco, alargamento do músculo cardíaco, batimento cardíaco mais forte, ECG anormal, frequência cardíaca e pulsação anormais;
- Coágulo na veia de um membro, choque;
- Dificuldades para respirar, distúrbios do trato respiratório, asma;
- Obstrução intestinal, nível elevado da enzima amilase, refluxo do conteúdo estomacal até a garganta, esvaziamento retardado do estômago;
- Inflamação da pele, sensação de queimação quando sob a luz solar;
- Distúrbios nas articulações;
- Menstruação dolorosa e sangramento menstrual anormal;
- Insuficiência de múltiplos órgãos, doença semelhante a gripe, sensibilidade elevada ao calor e ao frio, sensação de pressão no peito, agitação ou sensação anormal, perda de peso.
Reações adversas raras (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento)
- Pequenos sangramentos na pele devido a coágulos sanguíneos;
- Maior rigidez muscular;
- Cegueira, surdez;
- Acúmulo de líquido ao redor do coração;
- Falta de ar súbita;
- Formação de cisto no pâncreas;
- Problemas no fluxo sanguíneo do fígado;
- Doença grave com formação de bolhas na pele, boca, olhos e genitais; aumento dos pelos;
- Sede, quedas, sensação de pressão no peito, mobilidade reduzida, úlcera;
- Hepatite granulomatosa;
- Plexopatia braquial;
- Lesão do nervo periférico;
- Mudismo.
Reações adversas muito raras (ocorrem em menos de 0,01% dos pacientes que utilizam este medicamento)
- Fraqueza muscular;
- Exame cardíaco anormal;
- Insuficiência do fígado;
- Micção dolorosa com sangue na urina;
- Aumento do tecido gorduroso.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento.
Composição
Cápsula
Cada cápsula de 1 mg contém:
1 mg de tacrolimo.
Excipientes: Croscarmelose sódica, estearato de magnésio, lactose monoidratada, hipromelose, dióxido de titânio e gelatina.
Cada cápsula de 5 mg contém:
5 mg de tacrolimo.
Excipientes: Croscarmelose sódica, estearato de magnésio, lactose monoidratada, hipromelose, dióxido de titânio, óxido férrico vermelho e gelatina.
Solução injetável
Cada ampola de 1 mL contém:
5 mg de tacrolimo.
Excipientes: Álcool anidro (etanol) e óleo de rícino hidrogenado polioxietileno.
Superdosagem
Se você ingerir acidentalmente uma quantidade maior de Prograf, poderá apresentar as reações adversas mencionadas no item "Reações Adversas do Prograf".
Se isso acontecer, avise seu médico e ele recomendará o tratamento adequado.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800-722-6001, se você precisar de mais orientações.
Interação Medicamentosa
Devido ao potencial de insuficiência renal aditiva ou sinérgica, deve-se tomar cuidado ao administrar Tacrolimo monoidratado (substância ativa) com medicamentos que podem estar relacionados com disfunção renal. Esses medicamentos incluem, mas não estão limitados a, aminoglicosídeos, anfotericina B e cisplatina. Experimentos clínicos iniciais com a coadministração de Tacrolimo monoidratado (substância ativa) e ciclosporina resultaram em nefrotoxicidade aditiva/sinérgica.
Os pacientes que trocarem de ciclosporina para Tacrolimo monoidratado (substância ativa) não devem receber a primeira dose do mesmo antes de 24 horas depois da última dose de ciclosporina. A administração de Tacrolimo monoidratado (substância ativa) deve ser adiada na presença de níveis elevados de ciclosporina.
Fármacos que podem alterar as concentrações de tacrolimo
Como tacrolimo é metabolizado principalmente pelo sistema enzimático CYP3A, substâncias que inibem estas enzimas podem reduzir o metabolismo ou aumentar a biodisponibilidade de tacrolimo resultando em aumento nas concentrações plasmáticas ou no sangue total. Drogas que induzem estes sistemas enzimáticos podem aumentar o metabolismo ou diminuir a biodisponibilidade de tacrolimo, resultando em redução das concentrações no sangue total ou plasma. Monitoramento das concentrações sanguíneas e ajustes de dose são essenciais quando tais drogas são usadas concomitantemente:
Fármacos que podem aumentar as concentrações de tacrolimo no sangue:
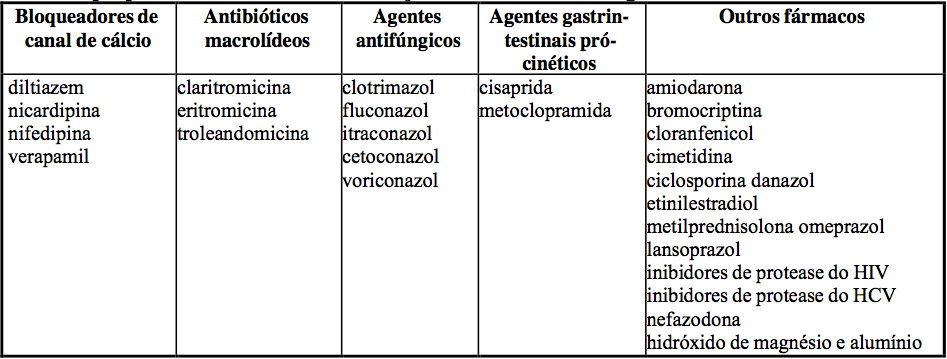
Em um estudo com 6 voluntários sadios, foi observado um aumento significante na biodisponibilidade oral do tacrolimo (de 14±5% para 30±8%) após o uso concomitante com cetoconazol (200 mg). A depuração aparente do tacrolimo administrado por via oral juntamente com cetoconazol diminuiu significativamente quando comparado com o tacrolimo administrado isoladamente (de 0,430±0,129 L/h/kg vs. 0,148±0,043 L/h/kg).
De modo geral, a administração por via IV não teve a depuração significativamente alterada pela coadministração com cetoconazol, no entanto houve uma grande variação entre os pacientes.
O lansoprazol (CYP2C19, substrato de CYP3A4) tem o potencial de inibir o metabolismo do tacrolimo mediado por CYP3A4 e, portanto, aumentar consideravelmente as concentrações de tacrolimo no sangue total, especialmente em pacientes transplantados que são metabolizadores deficientes ou intermediários de CYP2C19, em comparação aos pacientes que metabolizam CYP2C19 de forma eficiente.
A maioria dos inibidores de protease inibe as enzimas de CYP3A e pode aumentar as concentrações de tacrolimo no sangue total. Recomenda-se evitar o uso concomitante de tacrolimo com nelfinavir, a menos que os benefícios justifiquem os riscos. As concentrações de tacrolimo no sangue total são acentuadamente elevadas quando há coadministração de telaprevir ou boceprevir.
Recomenda-se o monitoramento das concentrações de tacrolimo no sangue total e das reações adversas associadas a tacrolimo, além de ajustes adequados no esquema de dose de tacrolimo quando tacrolimo e inibidores de protease (por exemplo, ritonavir, telaprevir, boceprevir) são usados concomitantemente.
Telaprevir
Em um estudo de dose única em 9 voluntários saudáveis, a coadministração de tacrolimo (dose única de 0,5 mg) com telaprevir (750 mg três vezes por dia por 13 dias) elevou a Cmáx de tacrolimo normalizado por dose em 9,3 vezes e a AUC em 70 vezes em comparação ao tacrolimo isolado.
Boceprevir
Em um estudo de dose única em 12 sujeitos de estudo, a coadministração de tacrolimo (dose única de 0,5 mg) com boceprevir (800 mg três vezes por dia por 11 dias) elevou a Cmáx de tacrolimo em 9,9 vezes e a AUC em 17 vezes em comparação ao tacrolimo em monoterapia.
Nelfinavir
Com base em um estudo clínico com 5 receptores de transplante hepático, a coadministração de tacrolimo e nelfinavir aumentou as concentrações de tacrolimo no sangue de forma significativa e, como resultado, uma redução de 16 vezes, em média, da dose de tacrolimo foi necessária para manter as concentrações médias de vale de tacrolimo de 9,7 ng/mL. Recomenda-se evitar o uso concomitante de Tacrolimo monoidratado (substância ativa) com nelfinavir, a menos que os benefícios justifiquem os riscos.
Fármacos que podem diminuir a concentração do tacrolimo no sangue:

A Erva de São João (Hypericum perforatum) induz o citocromo CYP3A4 e a glicoproteína P. Uma vez que o tacrolimo é substrato para o citocromo CYP3A4, há um potencial de que o uso da Erva de São João em pacientes recebendo Tacrolimo monoidratado (substância ativa) possa resultar na redução dos níveis de tacrolimo.
Em um estudo cruzado de dose única em pacientes sadios com coadministração oral de tacrolimo e hidróxido de alumínio e magnésio resultou em um aumento de 21% na AUC média do tacrolimo, e numa redução de 10% na Cmáx média de tacrolimo com relação a sua administração oral isolado.
Em um estudo com 6 voluntários normais observou-se uma significante redução na biodisponibilidade oral de tacrolimo (de 14±6% para 7±3%) quando administrado concomitantemente com rifampicina (600 mg). Além disso, houve um aumento significante da depuração do tacrolimo (de 0,036±0,008 L/h/kg para 0,053±0,010 L/h/kg) em administração concomitante com rifampicina.
Após a administração concomitante de tacrolimo e sirolimo (2 ou 5 mg/dia) em pacientes receptores de transplante renal estáveis, a AUC0-12 média e a Cmín reduziram em aproximadamente 30% com relação ao tacrolimo administrado isoladamente. Após a administração concomitante de tacrolimo e 1 mg/dia de sirolimo a
AUC0-12 média e a Cmín reduziram em aproximadamente 3% e 11%, respectivamente. A segurança e eficácia do uso do tacrolimo em combinação com o sirolimo para prevenção da rejeição a enxerto não foram estabelecidas, e seu uso não é recomendado.
Outras Interações Medicamentosas
Os imunossupressores podem afetar a vacinação. Portanto, durante o tratamento com Tacrolimo monoidratado (substância ativa), a vacinação pode ser menos eficaz. O uso de vacinas vivas deve ser evitado; vacinas vivas incluem, mas não são limitadas a sarampo, caxumba, rubéola, poliomielite, BCG, febre amarela e tifoide TY21a.
Ação da Substância
Resultados da eficácia
Transplante Hepático
A segurança e eficácia da imunossupressão baseada em Tacrolimo monoidratado (substância ativa) após transplante ortotópico de fígado foram avaliadas em dois estudos prospectivos, multicêntricos, abertos e randomizados. O grupo controle ativo foi tratado com regime de imunossupressão baseado em ciclosporina. Ambos os estudos utilizaram concomitantemente corticosteroides adrenais como parte do regime imunossupressor. Tais estudos foram desenhados com o objetivo de avaliar se os regimes imunossupressores eram equivalentes, tendo como desfecho primário a sobrevida de 12 meses após o transplante do paciente e do enxerto. A terapia de imunossupressão baseada em Tacrolimo monoidratado (substância ativa) se mostrou equivalente ao regime imunossupressor baseado em ciclosporina.
Em um ensaio envolvendo 529 pacientes em 12 centros nos Estados Unidos, antes da cirurgia 263 pacientes foram randomizados para o tratamento baseado em Tacrolimo monoidratado (substância ativa), enquanto 266 para o regime imunossupressor baseado em ciclosporina (CBIR). Em 10 dos 12 centros o mesmo protocolo de CBIR foi utilizado, enquanto 2 centros utilizaram protocolos diferentes.
Este ensaio clínico excluiu pacientes com disfunção renal, falência hepática fulminante com encefalopatia estágio IV e câncer. Foi permitida a inclusão de pacientes pediátricos (idade ? 12 anos).
Em um segundo ensaio clínico, 545 pacientes foram incluídos em 8 centros na Europa, antes da cirurgia 270 pacientes foram randomizados para o tratamento baseado em Tacrolimo monoidratado (substância ativa), enquanto 275 para CBIR. Neste estudo cada centro utilizou o próprio protocolo padrão de CBIR no braço controle-ativo. Não foram incluídos pacientes pediátricos, mas permitia a inclusão de indivíduos com disfunção renal, falência hepática fulminante com encefalopatia estágio IV e outros cânceres com metástases além do primário hepático.
As sobrevidas do paciente e do enxerto após 1 ano do transplante no grupo com regime imunossupressor baseado em Tacrolimo monoidratado (substância ativa) são equivalentes àquelas observadas nos grupos tratados com CBIR em ambos os estudos. A sobrevida geral do paciente (grupos recebendo regime imunossupressor baseado em Tacrolimo monoidratado (substância ativa) e CBIR combinados) foi de 88% no estudo americano e 78% no estudo europeu.
A sobrevida geral do enxerto após 1 ano do transplante (grupos recebendo regime imunossupressor baseado em Tacrolimo monoidratado (substância ativa) e CBIR combinados) foi de 81% no estudo americano e 73% no estudo europeu. Nos dois estudos a mediana de tempo de conversão da via de administração do Tacrolimo monoidratado (substância ativa) de IV para oral foi de 2 dias.
Devido a natureza e desenho dos estudos, a comparação de desfechos secundários, como incidência de rejeição aguda, rejeição refratária ou uso de OKT3 para rejeição esteroide-resistente, não pôde ser realizada adequadamente.
Transplante Renal
Foi realizado um estudo clínico de fase III, prospectivo, randomizado, aberto, multicêntrico, com imunossupressão baseada em Tacrolimo monoidratado (substância ativa) após transplante renal. Foram incluídos 412 pacientes receptores de transplante renal em 19 centros de estudo nos Estados Unidos. A terapia iniciou-se assim que a função renal foi estabelecida, como indicado pela creatinina sérica ? 4 mg/dL (mediana de 4 dias após o transplante, intervalo de 1 a 14 dias). Pacientes com menos de 6 anos de idade foram excluídos do ensaio.
Neste estudo foram incluídos 205 pacientes no grupo que recebeu imunossupressão baseada em Tacrolimo monoidratado (substância ativa), enquanto 207 pacientes foram randomizados para o grupo recebendo regime de imunossupressão com ciclosporina. Todos os pacientes receberam terapia de indução profilática, composta de uma preparação de anticorpos antilinfócito, corticosteroides e azatioprina.
As sobrevidas gerais de 1 ano dos pacientes e do enxerto foram de 96,1% e 89,6%, respectivamente, e foram equivalentes entre os dois tratamentos do estudo.
Devido a natureza do desenho dos estudos, a comparação de desfechos secundários, como incidência de rejeição aguda, rejeição refratária ou uso de OKT-3 para rejeição esteroide-resistente, não pôde ser realizada adequadamente.
Características Farmacológicas
Propriedades Farmacodinâmicas
O Tacrolimo monoidratado (substância ativa) prolonga a sobrevivência do hospedeiro e órgãos transplantados em modelos animais de transplantes de fígado, rins, coração, medula óssea, intestino delgado e pâncreas, pulmão e traqueia, pele, córnea e membros. Em animais, demonstrou-se que Tacrolimo monoidratado (substância ativa) causa supressão da imunidade humoral e, com maior extensão, as reações mediadas por células tais como a rejeição alográfica, hipersensibilidade do tipo tardia, artrite induzida por colágeno, encefalomielite alérgica experimental e doença do hospedeiro versus enxerto.
O Tacrolimo monoidratado (substância ativa) inibe a ativação do linfócito-T, apesar de seu exato mecanismo de ação não ser conhecido. Evidências experimentais sugerem que o Tacrolimo monoidratado (substância ativa) se liga a uma proteína intracelular, FKBP-12. Um complexo de Tacrolimo monoidratado (substância ativa)-FKBP-12, cálcio, calmodulina e calcineurina então se forma e a ação da fosfatase da calcineurina é inibida. Esse efeito pode impedir a desfosforilação e translocação do fator nuclear das células T-ativadas (NF-AT), um componente nuclear que inicia a transcrição genética para a formação de linfocinas (tais como interleucina-2, interferon gama). O resultado do mecanismo é a inibição da ativação do linfócito-T (isto é, imunossupressão).
Propriedades Farmacocinéticas
A atividade do Tacrolimo monoidratado (substância ativa) é primariamente devida ao fármaco. Os parâmetros farmacocinéticos do Tacrolimo monoidratado (substância ativa) foram determinados após administração intravenosa e oral em voluntários sadios, e em pacientes receptores de transplante renal e pacientes receptores de transplante hepático.
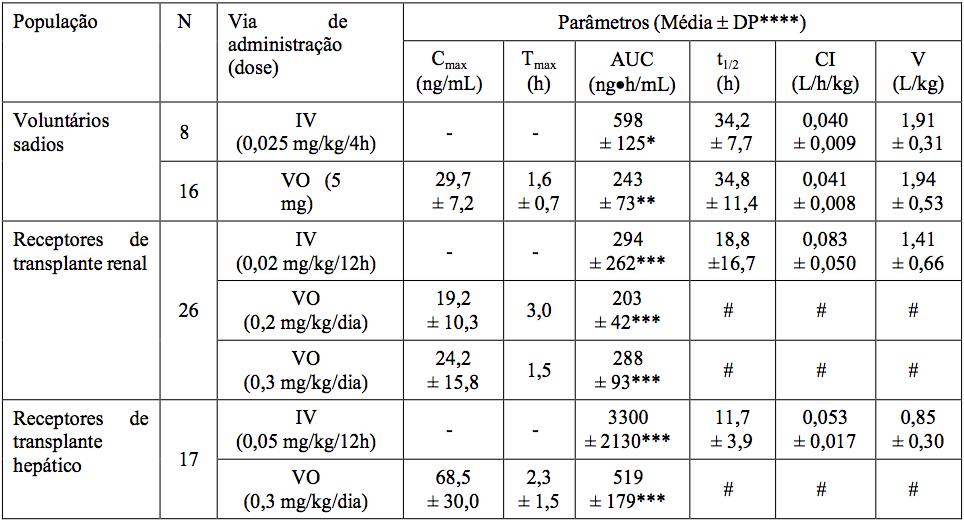
Corrigido para biodisponibilidade individual.
* AUC0-120.
**AUC0-72.
***AUC0-inf.
****DP.
= Desvio Padrão.
- Não aplicável.
# Dado indisponível.
Devido à variabilidade interindividual na farmacocinética do Tacrolimo monoidratado (substância ativa), é necessária a individualização da posologia para a otimização da terapia.
Os dados farmacocinéticos indicam que as concentrações no sangue total mais que as concentrações plasmáticas representam o compartimento de amostragem mais apropriado para descrever a farmacocinética do Tacrolimo monoidratado (substância ativa).
Absorção
A absorção de Tacrolimo monoidratado (substância ativa) a partir do trato gastrintestinal após a administração oral é incompleta e variável. A biodisponibilidade absoluta de Tacrolimo monoidratado (substância ativa) foi de 17±10% em pacientes adultos receptores de transplante renal (N=26), 22±6% em pacientes adultos receptores de transplante hepático (N=17) e 18±5% em voluntários sadios (N=16).
Um estudo com doses únicas conduzido em 32 voluntários sadios estabeleceu a bioequivalência de cápsulas com 1 mg e 5 mg. Em outro estudo com doses únicas em 32 voluntários sadios estabeleceu a bioequivalência entre as cápsulas de 0,5 mg e 1 mg de Tacrolimo monoidratado (substância ativa). A concentração máxima de Tacrolimo monoidratado (substância ativa) no sangue (Cmáx) e a área sob a curva (AUC) apresentaram um aumento proporcional à dose em 18 voluntários sadios em jejum que receberam uma dose única oral de 3,7 e 10 mg.
Em 18 pacientes receptores de transplante renal, concentrações mínimas de Tacrolimo monoidratado (substância ativa) de 3 a 30 ng/mL medidas 10 a 12 horas após a administração (Cmín) tiveram boa correlação com a AUC (coeficiente de correlação 0,93).
Em 24 pacientes receptores de transplante hepático em uma faixa de concentração de 10 a 60 ng/mL, o coeficiente de correlação foi 0,94.
Distribuição
A ligação de Tacrolimo monoidratado (substância ativa) às proteínas plasmáticas é de aproximadamente 99% e é independente da concentração dentro da faixa de 5 a 50 ng/mL.
O Tacrolimo monoidratado (substância ativa) é ligado principalmente à albumina e à alfa-1-glicoproteína ácida, e possui um elevado nível de associação com eritrócitos. A distribuição do Tacrolimo monoidratado (substância ativa) entre o sangue total e plasma depende de alguns fatores como hematócrito, temperatura no momento da separação do plasma, concentração do fármaco e a concentração de proteínas plasmáticas. Em um estudo norte-americano, a razão entre a concentração no sangue total e a concentração no plasma foi de 35 (intervalo de 12 a 67).
Metabolismo
O Tacrolimo monoidratado (substância ativa) é extensivamente metabolizado pelo sistema de oxidase de função mista, primariamente o sistema citocromo P-450 (CPY3A). Foi proposto um caminho metabólico que leva à formação de 8 metabólitos possíveis. A desmetilação e a hidroxilação foram identificadas como os mecanismos primários de biotransformação in vitro. O metabólito principal identificado em incubações com microssomas hepáticos humanos é o 13-desmetil Tacrolimo monoidratado (substância ativa). Em estudos in vitro, foi relatado que um metabólito 31-desmetil possui a mesma atividade do Tacrolimo monoidratado (substância ativa).
Excreção
A depuração média após administração intravenosa de Tacrolimo monoidratado (substância ativa) em voluntários sadios, pacientes adultos submetidos a transplante de rim e pacientes adultos submetidos a transplante de fígado é 0,040; 0,083 e 0,053 L/h/kg, respectivamente. Em humanos, menos de 1% da dose administrada foi excretada inalterada na urina. Em um estudo de balanço de massa com Tacrolimo monoidratado (substância ativa) radiomarcado administrado via intravenosa em 6 voluntários sadios, a recuperação média de material radiomarcado foi de 77,8±12,7%.
A eliminação fecal foi responsável por 92,4±1,0% e a meia-vida de eliminação baseada na radioatividade foi de 48,1±15,9 horas, enquanto que a meia-vida baseada na concentração de Tacrolimo monoidratado (substância ativa) foi 43,5±11,6 horas. A depuração média do Tacrolimo monoidratado (substância ativa) radiomarcado foi de 0,029±0,015 L/h/kg e a depuração média de Tacrolimo monoidratado (substância ativa) não marcado foi de 0,029±0,009 L/h/kg. Quando administrado via oral, a recuperação média de Tacrolimo monoidratado (substância ativa) radiomarcado foi 94,9±30,7%. A eliminação fecal foi responsável por 92,6±30,7%, a eliminação urinária por 2,3±1,1% e a meia-vida de eliminação baseada na radioatividade foi de 31,9±10,5 horas, enquanto que a baseada na concentração de Tacrolimo monoidratado (substância ativa) foi de 48,4±12,3 horas. A depuração média do Tacrolimo monoidratado (substância ativa) radiomarcado foi 0,226±0,116 L/h/kg e a depuração do Tacrolimo monoidratado (substância ativa) não marcado foi 0,172±0,088 L/h/kg.
Populações especiais
Pacientes Pediátricos
A farmacocinética de Tacrolimo monoidratado (substância ativa) foi estudada em pacientes receptores de transplante hepático, com idades entre 0,7 e 13,2 anos. Após administração via intravenosa de uma dose de 0,037 mg/kg/dia em 12 pacientes pediátricos, a meia-vida terminal média, o volume de distribuição médio e a depuração média foram de 11,5±3,8 horas, 2,6±2,1 L/kg e 0,138±0,071 L/h/kg, respectivamente.
Após administração oral em 9 pacientes, a AUC e a Cmáx médias foram 337±167 ng•h/mL e 43,4±27,9 ng/mL, respectivamente. A biodisponibilidade absoluta foi 31±21%.
As concentrações mínimas no sangue total de 31 pacientes com menos de 12 anos de idade mostraram que pacientes pediátricos necessitam de doses mais elevadas que os adultos para alcançar uma concentração mínima similar de Tacrolimo monoidratado (substância ativa).
Pacientes com Insuficiência Hepática e Renal
As médias dos parâmetros farmacocinéticos do Tacrolimo monoidratado (substância ativa), após administração única em pacientes com insuficiência hepática e renal, são dadas na seguinte tabela.
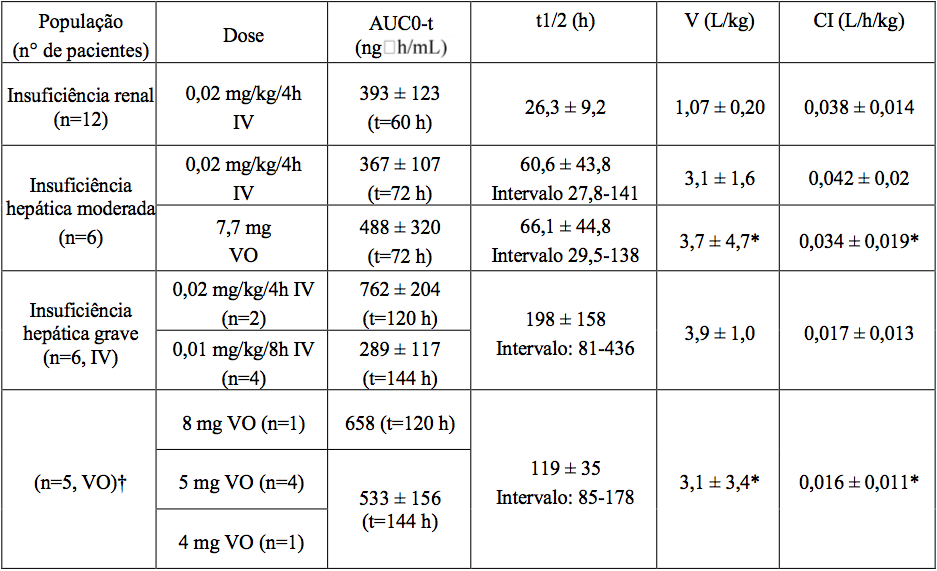 * corrigida para biodisponibilidade
* corrigida para biodisponibilidade
† 1 paciente não recebeu a dose por via oral
Pacientes com Insuficiência Renal
A farmacocinética do Tacrolimo monoidratado (substância ativa) após a administração de dose única intravenosa foi determinada em 12 pacientes (7 não estavam em diálise e 5 em diálise, creatinina sérica de 3,9±1,6 e 12,0±2,4 mg/dL, respectivamente) anteriormente ao transplante renal. Os parâmetros farmacocinéticos obtidos foram similares em ambos os grupos.
A depuração média de Tacrolimo monoidratado (substância ativa) em pacientes com disfunção renal foi similar a de voluntários normais.
Pacientes com Insuficiência Hepática
A farmacocinética do Tacrolimo monoidratado (substância ativa) foi determinada em 6 pacientes com leve disfunção hepática (escala Pugh média: 6,2) após administração de dose única via oral e intravenosa. A depuração média de Tacrolimo monoidratado (substância ativa) em pacientes com disfunção hepática leve não foi substancialmente diferente da depuração de voluntários normais. A farmacocinética do Tacrolimo monoidratado (substância ativa) foi estudada em 6 pacientes com disfunção hepática grave (média do escore de Pugh > 10). A média da depuração foi substancialmente menor nos pacientes com disfunção hepática, sem considerar a via de administração.
Raça
Não foi conduzido nenhum estudo formal para avaliar a disposição da farmacocinética do Tacrolimo monoidratado (substância ativa) em pacientes negros transplantados. No entanto, uma comparação retrospectiva entre pacientes negros e caucasianos receptores de transplante renal indicou que pacientes negros requerem doses mais altas de Tacrolimo monoidratado (substância ativa) para atingir concentrações mínimas similares.
Sexo
Não foi conduzido estudo formal para avaliar o efeito do sexo na farmacocinética de Tacrolimo monoidratado (substância ativa). No entanto, não se observa diferença na dosagem por sexo nos estudos clínicos envolvendo transplante renal. Uma comparação retrospectiva da farmacocinética em voluntários sadios, pacientes receptores de transplante renal e pacientes receptores de transplante hepático indicam que não há diferenças relacionadas ao sexo.
Cuidados de Armazenamento
Cápsula
Conserve as cápsulas de Prograf em temperatura ambiente (entre 15°C e 30°C), protegidas da umidade.
Este medicamento tem validade de 36 meses a partir da data de fabricação.
Depois de aberto, este medicamento pode ser utilizado em 12 meses.
Solução injetável
Prograf solução injetável deve ser armazenado em temperatura entre 5°C e 25°C, protegido da luz em sua embalagem original.
Este medicamento tem um prazo de validade de 24 meses após a data de fabricação.
Após preparo, manter Prograf solução injetável por 24 horas, armazenado em recipientes de vidro ou polietileno. Tacrolimo é absorvido por plástico PVC. Portanto, tubos, seringas e outros equipamentos usados para preparar, armazenar e administrar Prograf solução injetável não devem conter PVC.
Sob o ponto de vista microbiológico, o produto deve ser utilizado imediatamente após a diluição. Se não for utilizado imediatamente, o tempo e as condições armazenamento antes do uso são de responsabilidade do usuário e não deverão ser superiores a 24 horas entre 2 a 8°C, a menos que a diluição tenha ocorrido em condições assépticas controladas e validadas.
Número do lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido.
Guarde-o em sua embalagem original.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características organolépticas
Cápsula
Prograf 1 mg
Apresenta-se na forma de cápsula dura branca, com gravação em tinta vermelha de “1 mg” na tampa e “f 617” no corpo de cada cápsula.
Prograf 5 mg
Apresenta-se na forma de cápsula dura vermelho acinzentada, com gravação em tinta branca de “5 mg” na tampa e “f 657” no corpo de cada cápsula.
Solução injetável
Prograf solução injetável apresenta-se como um líquido claro e incolor, acondicionado em ampolas de vidro tipo I.
Número do lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido.
Guarde-o em sua embalagem original.
Todo medicamento deve ser mantido fora do alcance das crianças.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Dizeres Legais
MS-1.7717.0007.
Farmacêutico responsável:
Sandra Winarski
CRF-SP: 18.496.
Fabricado e embalado por:
Astellas Ireland Co. Ltd.
Killorglin – Co.
Kerry – Irlanda.
Registrado e importado por:
Astellas Farma Brasil Importação e Distribuição de Medicamentos Ltda.
Av. Guido Caloi, 1935, Bloco B, 2º andar
Santo Amaro, CEP: 05802-140
São Paulo – SP.
CNPJ 07.768.134/0001-04.
Venda sob prescrição médica.