Prolia 60mg/mL, caixa com 1 seringa preenchida com 1mL de solução de uso subcutâneo na Pharmed
Prolia 60Mg/Ml Solução Injetável 1 Seringa 1Ml

Prolia 60mg/mL, caixa com 1 seringa preenchida com 1mL de solução de uso subcutâneo na Pharmed
Não encontramos este produto para a Pharmed no Cliquefarma.
Confira os menores preços para Prolia 60Mg/Ml Solução Injetável 1 Seringa 1Ml
Prolia 60Mg/Ml Solução Injetável 1 Seringa 1Ml
30.63%
R$ 770,02
Agille Medicamentos
25.73%
R$ 824,40
Agille Speciality
25.73%
R$ 824,40
Mundial Farma
23.89%
R$ 844,80
Grupo Hera Medicamentos
23.87%
R$ 845,00
Para que serve
Prolia é indicado para tratar:
- Osteoporose em mulheres na fase de pós-menopausa. Nessas mulheres, Prolia aumenta a densidade mineral óssea (DMO) e reduz a incidência de fraturas de quadril, de fraturas vertebrais e não vertebrais.
- Perda óssea em pacientes submetidos a tratamentos de câncer de próstata ou de mama que causam diminuição hormonal. Nos pacientes com câncer de próstata, Prolia reduz a incidência de fraturas vertebrais.
- Osteoporose em homens.
Como o Prolia funciona?
Prolia contém denosumabe (substância ativa deste medicamento), uma proteína (chamada de anticorpo monoclonal) que interfere na ação de outra proteína a fim de tratar a perda óssea e a osteoporose.
O osso é um tecido vivo que se renova durante todo o tempo. O estrogênio é um hormônio que ajuda a manter os ossos saudáveis. Após a menopausa, os níveis de estrogênio caem, o que costuma tornar os ossos mais finos e frágeis. Isso pode às vezes levar a uma condição chamada de osteoporose. Muitas mulheres com osteoporose não apresentam sintomas, mas correm o risco de sofrer fraturas ósseas, especialmente na coluna, no quadril e nos punhos.
Cirurgias ou tratamentos medicamentosos de pacientes com câncer de próstata ou de mama que interrompem a produção de estrogênio ou de testosterona também podem levar à perda óssea. O osso se torna mais fraco e se quebra mais facilmente.
Seu médico fornecerá a você um cartão de lembrete do paciente, que contém informações de segurança importantes que você precisa saber antes e durante seu tratamento com Prolia.
Contraindicação
Este medicamento não deve ser utilizado caso você tenha hipocalcemia (baixa quantidade de cálcio no sangue).
Este medicamento não deve ser utilizado caso você apresente hipersensibilidade clinicamente significativa à denosumabe ou qualquer componente de Prolia.
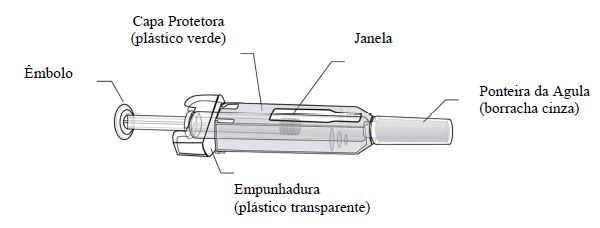
Como usar
Pessoas sensíveis ao látex não devem manusear a proteção da agulha da seringa preenchida descartável, que contém borracha natural seca (um derivado do látex).
Antes da administração, a solução de Prolia deve ser inspecionada para detecção de material particulado e de descoloração. A solução não deve ser usada se estiver turva ou com a coloração diferente da descrita no item " Cuidados de armazenamento do Prolia.
Não agite.
Para evitar desconforto no local da injeção, deixe a seringa preenchida atingir a temperatura ambiente (até 25°C) antes da aplicação. Injete lentamente todo o conteúdo da seringa preenchida. Descarte qualquer produto medicinal que permanecer na seringa preenchida.
As instruções de autoadministração por injeção subcutânea estão listadas a seguir.
Deve-se descartar qualquer produto não usado ou material residual, de acordo com as normas locais.
Incompatibilidades
Prolia não deve ser misturado com outros medicamentos.
Método de administração
A administração deve ser feita por uma pessoa adequadamente treinada em técnicas de injeção.
Seu médico prescreveu uma seringa preenchida de Prolia para injeção no tecido existente logo abaixo da pele (tecido subcutâneo). Você tem de injetar todo o conteúdo (1 mL) da seringa preenchida de Prolia, e a injeção deve ser aplicada uma vez a cada 6 meses, conforme as instruções do médico ou do profissional de saúde.
Equipamento
Para aplicar uma injeção, você precisará de:
- Uma seringa preenchida nova de Prolia;
- Um chumaço de algodão com álcool ou produto similar.
O que fazer antes de aplicar uma injeção subcutânea de Prolia
- Remova a seringa preenchida do refrigerador. NÃO pegue a seringa preenchida pelo êmbolo nem pela ponteira da agulha.
- NÃO aqueça a seringa de nenhuma outra maneira (nem no forno de micro-ondas nem em água quente). NÃO deixe a seringa exposta à luz direta.
- NÃO agite excessivamente a seringa preenchida.
- NÃO remova a ponteira da agulha da seringa preenchida até o momento da injeção.
- Verifique o prazo de validade no rótulo da seringa preenchida. NÃO use o produto se a data já tiver ultrapassado o último dia do mês impresso.
- Verifique a aparência de Prolia, que deve ser de uma solução transparente, incolor à ligeiramente amarelada. A solução não deve ser injetada se estiver turva ou com a coloração diferente da descrita acima.
- Escolha uma superfície confortável, bem iluminada e limpa, e coloque todo o equipamento ao alcance das mãos.
- Lave as mãos cuidadosamente.
Onde aplicar a injeção?
|
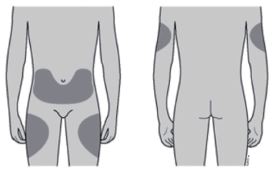
Os melhores locais para aplicar a injeção são a parte superior das coxas e o abdômen. A área externa dos braços também pode ser utilizada. |
|
Descarte de seringas usadas
- NÃO coloque a ponteira da agulha de volta nas seringas usadas.
- Mantenha as seringas usadas fora do alcance e da vista das crianças.
A seringa usada deve ser descartada de acordo com as normas locais. Pergunte ao farmacêutico como descartar medicamentos que já não são necessários. Essas medidas ajudarão a proteger o meio ambiente.
Instruções para injeção de Prolia com a seringa preenchida equipada com proteção manual da agulha
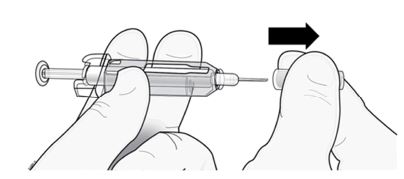
Importante: Para reduzir o risco de ferimentos acidentais com a agulha, a seringa preenchida descartável de Prolia tem uma capa de segurança verde; ative essa capa depois da aplicação da injeção.
Não mova a capa de segurança verde para a frente, ao longo da agulha, antes de administrar a injeção; o dispositivo travará e impedirá a aplicação da injeção.
Ative a capa de segurança verde (movendo-a ao longo da agulha) depois da administração da injeção.
A ponteira cinza da agulha da seringa preenchida descartável contém borracha natural seca (um derivado do látex). Pessoas sensíveis ao látex não devem manusear a ponteira.
1º passo: remova a ponteira cinza da agulha.
Remova a ponteira da agulha.
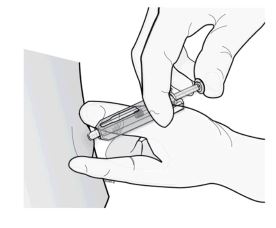
2º passo: aplique a injeção.
Insira a agulha e injete todo o líquido.
Não ponha a ponteira cinza de volta na agulha.
3° passo: mova imediatamente a capa de segurança verde ao longo da agulha.
Com a ponta da agulha voltada para a direção oposta à sua, segure a seringa preenchida pela empunhadura de plástico transparente com uma das mãos. Em seguida, com a outra mão, segure a capa de segurança verde pela base e mova-a suavemente em direção à agulha até que trave com firmeza e/ou você ouça um “clique”. Não segure a capa de segurança
verde com força – ela se moverá com facilidade se você a segurar e puxar suavemente.
Segure a empunhadura.
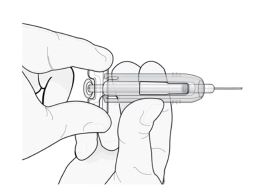
Mova suavemente a capa de segurança verde ao longo da agulha e trave-a com firmeza no lugar. Não segure a capa com muita força ao movê-la ao longo da agulha.
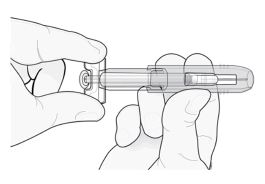
Descarte imediatamente a seringa e a ponteira da agulha no recipiente apropriado mais próximo. Não ponha a ponteira da agulha de volta na seringa usada.
Imediatamente descarte a seringa e a ponteira cinza da agulha de acordo com as normas locais. Não ponha a ponteira cinza de volta na agulha usada.
Posologia
A dose recomendada de Prolia é uma injeção subcutânea única de 60 mg administrada uma vez a cada 6 meses.
Os pacientes devem receber suplementos de cálcio e de vitamina D durante o tratamento.
Siga a orientação do seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu esquecer de usar Prolia?
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Converse com seu médico sobre a importância do consumo adequado de cálcio e de vitamina D durante o tratamento com Prolia.
A hipocalcemia (baixa quantidade de cálcio no sangue) precisa ser corrigida pela ingestão adequada de cálcio e de vitamina D antes do início do tratamento. O médico deve acompanhar os níveis de cálcio caso você tenha predisposição à hipocalcemia durante o tratamento, especialmente nas primeiras semanas do início da terapia (vide “QUAIS OS MALES QUE ESTE MEDICAMENTO PODE ME CAUSAR?”).
Num estudo clínico conduzido com mais de 7.800 mulheres em pós-menopausa com osteoporose, infecções graves que levaram à necessidade de internação foram relatadas com uma frequência maior no grupo de Prolia do que no grupo de placebo. Infecções graves na pele (0,1% placebo versus 0,4% Prolia) assim como infecções na região do abdômen (0,7% placebo versus 0,9% Prolia), do trato urinário (0,5% placebo versus 0,7% Prolia) e ouvido (0,0% placebo versus 0,1% Prolia) foram mais frequentes em pacientes tratados com Prolia. A inflamação de algumas regiões no coração (endocardite) também foi relatada mais frequentemente no grupo de pacientes tratados com Prolia (não houve ocorrência em pacientes recebendo placebo, mas houve relatos em 3 pacientes recebendo Prolia). A incidência de infecções que se aproveitam da queda da imunidade (oportunistas) foi semelhante entre os grupos placebo e Prolia e a incidência do total de infecções foi semelhante entre os grupos de tratamento.
Informe seu médico imediatamente caso você apresente dor, calor, vermelhidão e inchaço no local (infecção profunda ou superficial da pele).
Informe seu médico caso você tenha alergia ao látex (a proteção da agulha da seringa preenchida contém um derivado do látex).
Houve relatos de casos de osteonecrose de mandíbula (ONM), doença que atinge os vasos sanguíneos do osso, predominantemente em pacientes com câncer em estágio avançado que recebiam 120 mg de Prolia a cada 4 semanas. Também houve relatos, embora raros, de ONM em pacientes com osteoporose que tomavam 60 mg de Prolia a cada 6 meses (vide “QUAIS OS MALES QUE ESTE MEDICAMENTO PODE ME CAUSAR?”).
Higiene oral precária e procedimentos odontológicos invasivos (por exemplo, extração de dente) foram fatores de risco para ONM em pacientes recebendo Prolia em ensaios clínicos.
É importante avaliar os pacientes com fatores de risco para ONM antes de iniciar o tratamento. Se os fatores de risco forem identificados, é recomendado um exame dentário preventivo apropriado com dentista antes do tratamento com Prolia. Boas práticas de higiene oral devem ser mantidas durante o tratamento com Prolia. Se você usar dentaduras, você deve ter certeza de que elas encaixam apropriadamente. Caso você esteja em tratamento dentário ou será submetido à cirurgia dentária (por exemplo, extrações dentárias), informe seu médico sobre seu tratamento dentário e informe seu dentista que você está sendo tratado com Prolia.
Evitar procedimentos dentários invasivos durante o tratamento com Prolia. Para pacientes nos quais procedimentos dentários invasivos não podem ser evitados, o julgamento clínico do médico deverá orientar o plano de gestão de cada doente, com base na avaliação individual risco/benefício.
Os pacientes que são suspeitos de ter ou quem desenvolver ONM durante o uso de Prolia devem receber cuidados de um dentista ou um cirurgião-dentista. Em doentes que desenvolveram ONM durante o tratamento com Prolia uma interrupção temporária do tratamento deve ser considerada com base na avaliação individual do risco/benefício até que a condição seja resolvida.
Foram relatadas fraturas atípicas no fêmur (osso localizado na coxa) em pacientes recebendo Prolia. Essas fraturas podem ocorrer com mínimo ou nenhum trauma em algumas regiões do fêmur chamadas de subtrocanterianas ou diafisárias e podem ser bilaterais (em cada fêmur, nas duas coxas). Exames de RX da coxa caracterizam este evento. Também foram relatadas fraturas atípicas no fêmur em pacientes com certas doenças (por exemplo, deficiência de vitamina D, artrite reumatoide, hipofosfatasia) e com o uso de alguns medicamentos (por exemplo, bisfosfonatos, glicocorticoides, inibidores da bomba de prótons). Esses eventos também ocorreram sem terapia antirreabsortiva. Informe ao seu médico se durante o tratamento com Prolia você sentir dores novas ou incomuns na coxa, quadril ou virilha.
Em ensaios clínicos em mulheres com osteoporose pós-menopausa, o tratamento com Prolia resultou em significativa supressão de remodelação óssea, como evidenciado pelos marcadores de remodelação óssea e histomorfometria óssea. O significado destes resultados e o efeito do tratamento a longo prazo com Prolia são desconhecidos. As consequências a longo prazo do grau de supressão de remodelação óssea observado com Prolia pode contribuir para resultados adversos, como osteonecrose de mandíbula, fraturas atípicas e consolidação da fratura atrasada. Os pacientes devem ser monitorados para estas consequências.
Fraturas vertebrais múltiplas (FVM) podem ocorrer após descontinuação de tratamento com Prolia, particularmente em pacientes com um histórico de fratura vertebral.
Você não deve interromper a terapia com Prolia sem a orientação do seu médico. Seu médico avaliará o risco/benefício individual antes de descontinuar o tratamento com Prolia. Caso o tratamento com Prolia seja descontinuado, fale com seu médico sobre a transição para uma terapia alternativa a fim de tratar perda óssea e osteoporose.
Reações Adversas
Reações muito comuns (ocorrem em mais de 10% dos pacientes que utilizam este medicamento)
Artralgia2, dor nas costas.
Reações comuns (ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento)
Catarata2, dor nas extremidades, cistite, infecção do trato respiratório superior, pneumonia, faringite, herpes zoster, anemia, angina de peito, fibrilação atrial, hipercolesterolemia, rash, prurido, dor musculoesquelética, dor óssea, mialgia, osteoartrite espinal, dor abdominal superior, flatulência, refluxo gastroesofágico, edema periférico, astenia, dor ciática, vertigem, insônia.
Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento)
Infecção profunda ou superficial da pele (celulite1), irritação da pele3 (eczema).
Reações raras (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento)
Osteonecrose de mandíbula (doença que acomete os vasos sanguíneos do osso), hipocalcemia1 (baixa quantidade de cálcio no sangue).
Reações muito raras (ocorrem em menos de 0,01% dos pacientes que utilizam este medicamento)
Fratura femoral atípica1,4 (fraturas em locais do fêmur relacionadas a mínimo ou nenhum trauma).
1 Vide Precauções do Prolia.
2 Nos homens com câncer de próstata sob terapia de privação androgênica.
3 Inclui dermatite (inflamação da pele), dermatite alérgica, dermatite atópica e dermatite de contato.
4 No programa de estudos clínicos de osteoporose foram relatadas fraturas femorais atípicas em pacientes tratados com Prolia.
Infecções graves
Em um estudo clínico com 7.808 mulheres na pós-menopausa com osteoporose, a incidência de infecções resultantes em morte foi de 0,2% em ambos os grupos (placebo e tratados com Prolia). Entretanto, a incidência de infecções graves não fatais foi de 3,3% no grupo placebo e 4,0% nos grupos Prolia. Foram relatadas internações por infecções graves no abdômen (0,7% placebo versus 0,9% Prolia), do trato urinário (0,5% placebo versus 0,7% Prolia) e ouvido (0,0% placebo versus 0,1% Prolia). Endocardite não foi relatada em pacientes com placebo e houve relato em três pacientes que receberam Prolia. As infecções de pele, incluindo erisipela e celulite, levando à hospitalização foram relatadas com maior frequência em
pacientes tratados com Prolia (0,1% placebo versus 0,4% Prolia). A incidência de infecções oportunistas foi similar a reportada com placebo.
Pancreatite
A pancreatite foi relatada em 4 pacientes (0,1%) no grupo placebo e 8 pacientes (0,2%) no grupo Prolia. Desses relatos, 1 paciente do grupo placebo e todos os 8 pacientes do grupo Prolia tiveram eventos graves, incluindo um óbito no grupo Prolia. Vários pacientes tinham antecedentes de pancreatite. O tempo de administração do produto até a ocorrência do evento foi variável.
Novas doenças malignas
A incidência global de novas doenças malignas foi de 4,3% no grupo placebo e 4,8% no grupo Prolia. Novos tumores malignos relacionados com a mama (0,7% placebo versus 0,9% Prolia), sistema reprodutor (0,2% placebo versus 0,5% Prolia), e do sistema gastrointestinal (0,6% placebo versus 0,9% Prolia) foram relatados. A relação causal com a exposição à droga ainda não foi estabelecida.
Imunogenicidade
O denosumabe é um anticorpo monoclonal humano. Como ocorre com todas as proteínas terapêuticas, apresenta potencial teórico de imunogenicidade. Mais de 13.000 pacientes foram selecionados para receber anticorpos de ligação, usando-se um imunoensaio de comparabilidade de eletroquimioluminescência. Menos de 1% dos pacientes tratados com o denosumabe por
até 5 anos apresentaram resultados positivos (incluindo anticorpos preexistentes, transitórios e em desenvolvimento). Naqueles que tiveram resultados positivos para anticorpos de ligação também se avaliaram os anticorpos neutralizantes, usando-se um ensaio biológico quimioluminescente in vitro com análise de células. Nenhum dos pacientes apresentou resultado positivo.
Nenhum indício de alteração do perfil farmacocinético, do perfil de toxicidade ou da resposta clínica foi associado com o desenvolvimento de anticorpos de ligação.
Osteonecrose de mandíbula
ONM foi relatada raramente, em 14 pacientes, em estudos clínicos em pacientes com osteoporose e câncer de mama ou próstata recebendo ablação de hormônio, incluindo um total de 19.521 pacientes.
Fratura femoral atípica
No programa de estudo clínico de osteoporose, as fraturas femorais atípicas foram relatadas em pacientes tratados com Prolia.
Fraturas vertebrais múltiplas (FVM) após descontinuação do tratamento com Prolia
No programa de estudo clínico de osteoporose, FVM foram relatadas em pacientes após a descontinuação do tratamento com Prolia, particularmente naqueles com um histórico de fratura vertebral.
Dados pós-comercialização
Reações de hipersensibilidade
Reações de hipersensibilidade relacionadas ao medicamento, incluindo erupção na pele, inchaço facial, eritema (vermelhidão na pele) e reações anafiláticas (reações alérgicas graves), foram relatadas em pacientes recebendo Prolia.
Hipocalcemia grave
Foi relatada hipocalcemia (baixa quantidade de cálcio no sangue) sintomática grave em pacientes recebendo Prolia que se encontravam sob risco aumentado de hipocalcemia.
Dor musculoesquelética
Foi relatada dor musculoesquelética, incluindo casos graves, em pacientes recebendo Prolia.
Hiperparatireoidismo
Hormônio da paratireoide (PTH): elevação acentuada do PTH tem sido observada em pacientes com insuficiência renal grave (clearance de creatinina < 30 mL/min) ou em diálise.
Atenção: Este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista. Informe também à empresa através do seu serviço de atendimento.
População Especial
Crianças
Não se recomenda o uso de Prolia para pacientes pediátricos porque a segurança e a eficácia deste medicamento não foram estabelecidas para esse grupo de pacientes.
Idosos
Com base nos dados disponíveis de segurança e eficácia, concluiu-se que não é necessário nenhum ajuste de dose em idosos.
Insuficiência renal
Com base nos dados disponíveis de segurança e eficácia, concluiu-se que não é necessário nenhum ajuste de dose para pacientes com insuficiência renal.
Os pacientes com insuficiência renal grave ou que se submetem a diálise correm maior risco de desenvolver hipocalcemia. A ingestão adequada de cálcio e de vitamina D é importante nesses casos.
Insuficiência hepática
A segurança e a eficácia de Prolia para os pacientes com insuficiência hepática não foram estudadas.
Gravidez
Não há dados disponíveis sobre o efeito deste medicamento na gravidez, portanto não se recomenda o uso de Prolia para mulheres grávidas.
Lactação
Não se sabe se o denosumabe é excretado no leite humano. Como este medicamento tem o potencial de causar reações adversas em bebês que são amamentados, o médico deve avaliar a interrupção do aleitamento ou do uso de Prolia.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Medicamentos com o Mesmo Princípio Ativo
Prolia contém o mesmo princípio ativo (denosumabe) encontrado em XGEVA®. Pacientes recebendo Prolia não devem receber XGEVA.
Efeitos sobre a capacidade de dirigir veículos e de operar máquinas
Não houve estudos sobre o efeito na capacidade de dirigir veículos ou de operar máquinas pesadas em pacientes sob tratamento com denosumabe.
Interações medicamentosas
Prolia não afeta a farmacocinética do medicamento midazolam, que é metabolizado por uma enzima chamada de citocromo P450 34A (CYP34A) o que indica que Prolia não deve afetar a farmacocinética de medicamentos metabolizados por essa enzima. Sendo assim não deve existir interferência de Prolia no funcionamento de outro medicamento e vice-versa.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento. É especialmente importante que você informe ao seu médico se você está sendo tratado com outro medicamento contendo denosumabe.
Você não deve administrar Prolia com outro medicamento contendo denosumabe.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Composição
Apresentação
Solução injetável 60 mg/mL em embalagem com 1 seringa preenchida de 1,0 mL.
USO SUBCUTÂNEO
USO ADULTO ACIMA DE 18 ANOS
Composição
Cada 1,0 mL contém:
|
60 mg/mL | |
|
Denosumabe |
60 mg |
|
Excipientes: ácido acético glacial, hidróxido de sódio, sorbitol, polissorbato 20 e água para injetáveis. |
q.s.p. |
Superdosagem
Não há dados disponíveis de estudos clínicos sobre superdosagem de Prolia.
No entanto, em estudos clínicos preliminares denosumabe foi administrado em doses maiores, de até 180mg a cada 4 semanas (doses cumulativas de até 1.080 mg durante 6 meses), representando 18 vezes a posologia recomendada para o tratamento da osteoporose (60 mg a cada 6 meses).
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento se possível. Ligue para 0800 722 6001 se você precisar de mais orientações.
Interação Medicamentosa
Denosumabe (substância ativa) não afeta a farmacocinética de midazolam, que é metabolizado pelo citocromo P450 34A (CYP34A) o que indica que Denosumabe (substância ativa) não deve afetar a farmacocinética de medicamentos metabolizados por essa enzima.
Ação da Substância
Resultados de eficácia
Tratamento da osteoporose pós-menopáusica
A eficácia e a segurança do Denosumabe (substância ativa) no tratamento de osteoporose pós-menopáusica foram demonstradas no Freedom, um estudo multinacional randomizado, duplo-cego, controlado com placebo, com duração de 3 anos. O estudo demonstrou que o Denosumabe (substância ativa) foi eficaz, em comparação com placebo, na redução de novas fraturas vertebrais, não vertebrais e de quadril em mulheres na fase de pós-menopausa com osteoporose. Foram recrutadas 7.808 mulheres com 60 a 91 anos de idade, das quais 23,6% tinham principalmente fraturas vertebrais.
As mulheres foram randomizadas para receber injeções subcutâneas de placebo (n = 3.906) ou Denosumabe (substância ativa) 60 mg (n = 3.902) uma vez a cada 6 meses.
Receberam suplementos diários de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 400 UI). A variável de eficácia primária foi a incidência de novas fraturas vertebrais. As variáveis de eficácia secundárias incluíram a incidência de fraturas não vertebrais e de quadril, avaliadas após 3 anos.
O Denosumabe (substância ativa) reduziu significativamente o risco de novas fraturas vertebrais, não vertebrais e de quadril, em comparação com placebo. Os 3 desfechos de eficácia em fraturas atingiram o nível de significância estatística (p < 0,05), com base no esquema predefinido de testes sequenciais.
Efeito sobre fraturas vertebrais
O Denosumabe (substância ativa) reduziu significativamente o risco de novas fraturas vertebrais (desfecho primário), em 68% (relação de risco de 0,32; p < 0,0001), em 3 anos.
As taxas dessas fraturas foram de 7,2% no grupo de placebo e 2,3% no de Denosumabe (substância ativa) (redução de risco absoluto não ajustada de 4,8%). Também se observaram as reduções obtidas em 1 ano (redução de risco relativo de 61%; redução de risco absoluto não ajustada de 1,4%) e em 2 anos (redução de risco relativo de 71%; redução de risco absoluto não ajustada de 3,5%) (para todos, p < 0,0001).
O Denosumabe (substância ativa) também reduziu, em 3 anos, o risco de outras categorias de fratura predefinidas. São elas: novas fraturas vertebrais ou agravamento de fraturas vertebrais (redução do risco relativo de 67%, redução do risco absoluto não ajustada de 4,8%), novas fraturas vertebrais múltiplas (redução do risco relativo de 61%, redução do risco absoluto não ajustada de 1,0%) e fraturas vertebrais clínicas (redução do risco relativo de 69%, redução do risco absoluto não ajustada de 1,8%).
As reduções do risco de novas fraturas vertebrais pelo Denosumabe (substância ativa) durante 3 anos foram persistentes e significativas, independentemente do risco basal de fraturas osteoporóticas graves em 10 anos, avaliado pelo Frax(algoritmo de avaliação do risco de fraturas da OMS), e do fato de as mulheres terem fratura vertebral prevalente ou histórico de fratura não vertebral. A idade na avaliação basal, a DMO, a remodelação óssea e o uso prévio de produto medicinal para osteoporose também não influíram nessas reduções.
Em mulheres na fase de pós-menopausa e com mais de 75 anos, o Denosumabe (substância ativa) reduziu a incidência de novas fraturas vertebrais (64%) e não vertebrais (16%).
Efeito sobre todas as fraturas clínicas
O Denosumabe (substância ativa) reduziu significativamente o risco de fraturas não vertebrais (desfecho secundário), em 20% (razão de risco de 0,80; p = 0,0106), em 3 anos. As taxas dessas fraturas foram de 8,0% no grupo de placebo e 6,5% no de Denosumabe (substância ativa) (redução do risco absoluto não ajustada de 1,5%). Este medicamento também diminuiu o risco de fraturas clínicas (redução de risco relativo de 30%, redução do risco absoluto não ajustada de 2,9%), fraturas não vertebrais graves (redução de risco relativo de 20%, redução do risco absoluto não ajustada de 1,2%) e fraturas osteoporóticas graves (redução de risco relativo de 35%, redução do risco absoluto não ajustada de 2,7%) em 3 anos.
Em mulheres com pontuação T basal de DMO do colo do fêmur ? -2,5, o Denosumabe (substância ativa) reduziu a incidência de fraturas não vertebrais (redução de risco relativo de 35%, redução do risco absoluto não ajustada de 4,1%, p < 0,001) em 3 anos. Tais reduções ocorreram independentemente de ter sido observada à entrada no estudo a probabilidade de ocorrência de fratura osteoporótica grave em 10 anos, de acordo com o Frax.
Efeito sobre as fraturas de quadril
O Denosumabe (substância ativa) reduziu significativamente o risco de fraturas de quadril (desfecho secundário), em 40% (razão de risco de 0,60; p = 0,0362), em 3 anos. As taxas dessas fraturas foram de 1,2% no grupo de placebo e de 0,7% no de Denosumabe (substância ativa) (redução do risco absoluto não ajustada de 0,5%). Tais reduções foram constantes e significativas e ocorreram independentemente de ter sido observada à entrada no estudo a probabilidade de ocorrência de fratura de quadril em 10 anos, de acordo com o Frax.
Em mulheres com alto risco de fraturas, conforme definido acima por idade basal, DMO e fratura vertebral prevalente, observou-se redução de 48% no risco relativo com o Denosumabe (substância ativa) (redução do risco absoluto não ajustada de 1,1%).
Em análise post-hoc em mulheres na fase de pós-menopausa, com osteoporose e idade acima de 75 anos, o Denosumabe (substância ativa) reduziu a incidência de fraturas de quadril (62%).
Efeito sobre a densidade mineral óssea (DMO)
O Denosumabe (substância ativa) aumentou significativamente a DMO, em relação ao tratamento com placebo, em todos os locais clínicos medidos após 1, 2 e 3 anos. O aumento em 3 anos foi de 9,2% na coluna lombar, 6,0% no quadril, 4,8% no colo do fêmur, 7,9% no trocânter no quadril, 3,5% no terço distal do rádio e 4,1% no corpo inteiro. Na DMO da coluna lombar, do quadril e do trocânter do quadril, a alteração foi observada no período de um mês após a dose inicial. O Denosumabe (substância ativa) aumentou a DMO da coluna lombar em relação à avaliação basal em 96% em mulheres pós menopáusicas após 3 anos. Observaram-se efeitos consistentes sobre a DMO da coluna lombar, independentemente de idade basal, raça, peso/IMC, DMO e nível de remodelação óssea.
Histologia óssea
As avaliações da histologia mostraram ossos com arquitetura e qualidade normais, bem como a redução esperada de remodelação óssea em relação ao tratamento com placebo. Não houve evidências de defeitos de mineralização, osso trançado (não lamelar) ou fibrose de medula.
Estudo de extensão aberto no tratamento de osteoporose pós-menopáusica
Um total de 4.550 pacientes que concluíram o estudo Freedom (N = 7.808) foi recrutado para um estudo de extensão multinacional, multicêntrico, aberto e de braço único, com 7 anos de duração, conduzido para avaliar a segurança e a eficácia em longo prazo de Denosumabe (substância ativa). Todos os pacientes no estudo de extensão receberam Denosumabe (substância ativa) a cada 6 meses em dose única de 60 mg por via subcutânea, bem como suplemento diário de cálcio (pelo menos 1 g) e vitamina D (pelo menos 400 UI).
Com base nos dados obtidos nos primeiros dois anos do estudo de extensão referentes aos pacientes que receberam Denosumabe (substância ativa) no estudo Freedom e continuaram em tratamento (anos 4 e 5 de tratamento com Denosumabe (substância ativa)), a taxa geral de incidência de eventos adversos e de eventos adversos sérios relatados foi semelhante à observada nos primeiros três anos do estudo Freedom.
Para os pacientes que passaram a receber Denosumabe (substância ativa) após placebo no estudo Freedom, a taxa geral de incidência de eventos adversos e de eventos adversos sérios relatados também foi semelhante à observada nos primeiros três anos do estudo Freedom. Foram observados dois casos de osteonecrose de mandíbula, ambos solucionados.
O tratamento com Denosumabe (substância ativa) manteve baixa incidência de novas fraturas vertebrais e não vertebrais nos anos 4 e 5 (2,8% dos pacientes apresentaram pelo menos uma nova fratura vertebral até o mês 24, 2,5% dos pacientes apresentaram fratura não vertebral).
O tratamento com Denosumabe (substância ativa) continuou a aumentar a DMO na coluna lombar (3,3%), quadril total (1,3%), colo femoral (1,2%) e trocânter (1,8%) nos anos 4 e 5. O aumento percentual na DMO em relação ao período inicial do estudo original Freedom (ou seja, após 5 anos de tratamento), no grupo em longo prazo, foi de 13,8% na coluna lombar, 7,0% no quadril total, 6,2% no colo femoral e 9,7% no trocânter.
Dados clínicos comparativos com os de alendronato em mulheres pós-menopáusicas com baixa massa óssea
Em 2 estudos randomizados, duplo-cegos e controlados com ativo, um em mulheres não submetidas a tratamento anterior e outro em mulheres previamente tratadas com alendronato, o Denosumabe (substância ativa) mostrou aumentos significativamente maiores da DMO e reduções dos marcadores de remodelação óssea (por exemplo, CTX sérico), em comparação com alendronato.
Observaram-se aumentos consistentemente maiores da DMO de coluna lombar, quadril, colo do fêmur, trocânter e terço distal do rádio em mulheres tratadas com o Denosumabe (substância ativa), em comparação com as que continuaram a receber alendronato (para todos, p < 0,05).
Eficácia clínica do tratamento da perda óssea associada com ablação hormonal
Tratamento da perda óssea associada com privação androgênica
A eficácia e a segurança do Denosumabe (substância ativa) no tratamento da perda óssea associada com privação androgênica foram avaliadas em um estudo multinacional randomizado, duplo-cego, controlado com placebo, com duração de 3 anos, em 1.468 homens com câncer de próstata não metastático e idade de 48 a 97 anos.
Os homens com menos de 70 anos também tinham pontuação T de DMO da coluna lombar, do quadril ou do colo do fêmur < -1,0 ou histórico de fratura osteoporótica.
Os pacientes receberam injeções subcutâneas de Denosumabe (substância ativa) 60 mg (n = 734) ou placebo (n = 734) uma vez a cada 6 meses, além de suplementos diários de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 400 UI).
Observaram-se aumentos significativos da DMO de coluna lombar, quadril e colo do fêmur e trocânter do quadril no período de 1 mês após a dose inicial. O Denosumabe (substância ativa) aumentou a DMO da coluna lombar em 7,9%, do quadril em 5,7%, do colo do fêmur em 4,9%, do trocânter do quadril em 6,9%, do terço distal do rádio em 6,9% e do corpo inteiro em 4,7%, no período de 3 anos, em relação a placebo (p < 0,0001).
Os efeitos do tratamento sobre a DMO da coluna lombar foram persistentes independentemente dos dados basais relativos a idade, raça, região geográfica, peso/IMC, DMO, nível de remodelação óssea, duração da privação androgênica e presença de fratura vertebral.
O Denosumabe (substância ativa) reduziu significativamente o risco de novas fraturas vertebrais, em 62% (razão de risco de 0,38; p < 0,0063), em 3 anos. Também foram observadas as reduções obtidas em 1 ano (redução do risco relativo de 85%; redução do risco absoluto de 1,6%) e 2 anos (redução do risco relativo de 69%; redução do risco absoluto de 2,2%) (para todos, p < 0,01). Este medicamento reduziu ainda em 72% a incidência de mais de uma fratura osteoporótica em um mesmo paciente, em qualquer local, em 3 anos, com relação a placebo (taxa de 2,5% com placebo versus 0,7% com Denosumabe (substância ativa); p = 0,0063).
Tratamento da perda óssea em mulheres sob tratamento com inibidores da aromatase para câncer de mama
A eficácia e a segurança do Denosumabe (substância ativa) no tratamento da perda óssea decorrente da terapia adjuvante com inibidores da aromatase foram avaliadas em um estudo multinacional randomizado, duplo-cego, controlado com placebo, com duração de 2 anos, em 252 mulheres com câncer de mama não metastático e idade de 35 a 84 anos. As pacientes tinham pontuações T basais de DMO de -1,0 a -2,5 na coluna lombar, no quadril ou no colo do fêmur.
Foram randomizadas para receber injeções subcutâneas de Denosumabe (substância ativa) 60 mg (n = 127) ou placebo (n = 125) uma vez a cada 6 meses. Receberam suplementos diários de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 400 UI). A variável de eficácia primária foi a alteração percentual da DMO da coluna lombar.
O Denosumabe (substância ativa) aumentou significativamente a DMO em todos os locais clínicos medidos, em relação ao tratamento com placebo, após 2 anos: 7,6% na coluna lombar, 4,7% no quadril, 3,6% no colo do fêmur, 5,9% no trocânter do quadril, 6,1% no terço distal do rádio e 4,2% no corpo inteiro. Os aumentos da DMO da coluna lombar já foram significativos 1 mês após a dose inicial.
Os efeitos do tratamento sobre a DMO da coluna lombar foram persistentes independentemente de idade inicial, duração da terapia com inibidores da aromatase, peso/IMC, quimioterapia prévia, uso prévio de moduladores dos receptores seletivos de estrogênio (SERM) e tempo desde a menopausa.
Tratamento da osteoporose em homens
A eficácia e a segurança do Denosumabe (substância ativa) no tratamento de homens com osteoporose foram demonstradas em um estudo multinacional, controlado com placebo, duplo-cego, randomizado, com duração de 1 ano, em homens com baixa massa óssea, que apresentaram pontuação T basal de DMO entre -2,0 e -3,5 na coluna lombar ou no colo do fêmur. Homens com pontuação T basal de DMO entre -1,0 e -3,5 na coluna lombar ou no colo do fêmur e com histórico de fratura anterior por fragilidade também foram inscritos. Homens com outras doenças (tais como artrite reumatoide, osteogênese imperfeita e doença de Paget) ou em terapias capazes de afetar os ossos foram excluídos deste estudo.
Os 242 homens inscritos no estudo tinham idades na faixa de 31 a 84 anos e foram randomizados para receber injeções subcutâneas de placebo (n = 121) ou de Denosumabe (substância ativa) 60 mg (n = 121) uma vez a cada 6 meses. Os pacientes também receberam pelo menos 1.000 mg de cálcio e pelo menos 800 UI de suplemento de vitamina D diariamente.
A variável primária de eficácia foi o percentual de mudança na DMO da coluna lombar em 1 ano. As variáveis secundárias de eficácia incluíram o percentual de mudança na DMO do quadril total, do trocânter do quadril, do colo do fêmur e do terço do rádio distal em 1 ano e mudança no telopeptídeo C (CTX) no dia 15.
O tratamento com Denosumabe (substância ativa) aumentou significativamente a DMO a partir do momento basal na coluna lombar e em todas as regiões esqueléticas (fêmur proximal, rádio distal) em 1 ano. O Denosumabe (substância ativa) aumentou a DMO da coluna lombar em 4,8%, a DMO do quadril total em 2,0%, do trocânter do quadril em 2,3%, a DMO do colo do fêmur em 2,2% e a DMO do terço rádio distal em 0,9%, em comparação com o placebo.
Aumentos na DMO da coluna lombar, do quadril total e do trocânter do quadril foram observados em até 6 meses. Denosumabe (substância ativa) aumentou a DMO da coluna lombar a partir do momento basal em 94,7% dos homens em 1 ano.
Foram observados efeitos consistentes na DMO da coluna lombar independentemente da idade, da raça, do peso/índice de massa corporal (IMC), da DMO e de remodelação óssea.
Histologia e histomorfometria do osso
Foi obtido um total de 29 amostras de biópsia da crista óssea da região transilíaca de homens com osteoporose em 12 meses (17 amostras do grupo Denosumabe (substância ativa), 12 amostras do grupo placebo). As avaliações histológicas qualitativas mostraram arquitetura normal e qualidade sem evidências de defeitos de mineralização, de osso não lamear ou de fibrose medular em pacientes tratados com Denosumabe (substância ativa).
Características farmacológicas
Farmacodinâmica
Mecanismo de ação
O Denosumabe (substância ativa) é um anticorpo monoclonal humano (IgG2) que tem como alvo o RANKL, ao qual se liga com grande afinidade e especificidade, impedindo que o ligante ative seu único receptor, o RANK, na superfície dos osteoclastos e seus precursores, independentemente da superfície óssea. A prevenção da interação RANKL/RANK inibe a formação, a função e a sobrevivência de osteoclastos. O Denosumabe (substância ativa), portanto, reduz a reabsorção óssea e aumenta a massa e a resistência dos ossos corticais e trabeculares.
Efeitos farmacodinâmicos
Em estudos clínicos, o tratamento com 60 mg de Denosumabe (substância ativa) resultou em rápida redução (de aproximadamente 70%) do marcador de reabsorção óssea tipo 1 no soro, o telopeptídeo C (CTX), no período de 6 horas após a administração subcutânea, atingindo-se cerca de 85% de redução em 3 dias. As reduções do CTX se mantiveram ao longo do intervalo de administração de 6 meses. Ao final de cada intervalo, eram parcialmente atenuadas, com máxima ? 87% e mínima ? 45% (faixa de 45% a 80%), o que reflete a reversibilidade dos efeitos do Denosumabe (substância ativa) sobre a remodelação óssea assim que os níveis séricos diminuem. Esses efeitos foram mantidos com a continuação do tratamento. De maneira condizente com o acoplamento fisiológico de formação e reabsorção óssea na remodelação esquelética, observaram-se reduções dos marcadores de formação óssea (por exemplo, fosfatase alcalina específica dos ossos [BSAP] e propetídeo N-terminal sérico do colágeno de tipo 1 [P1NP]), iniciadas 1 mês após a primeira dose do Denosumabe (substância ativa).
De modo geral, os marcadores de remodelação óssea (marcadores de reabsorção e formação óssea) atingiram os níveis pré-tratamento no período de 9 meses após a última dose subcutânea de 60 mg. A cada retomada do tratamento, o grau de inibição de CTX obtido com o Denosumabe (substância ativa) foi similar ao observado no uso inicial desse medicamento.
Em um estudo clínico em mulheres pós-menopáusicas com baixa massa óssea (N = 504) que haviam recebido alendronato pelo período mediano de 3 anos, as pacientes que passaram a ser tratadas com Denosumabe (substância ativa) apresentaram reduções adicionais do CTX sérico, em comparação às que continuaram recebendo alendronato. Nesse estudo, as alterações dos níveis séricos de cálcio foram similares entre os 2 grupos.
Farmacocinética
Após a administração subcutânea, o Denosumabe (substância ativa) exibiu farmacocinética não linear com as doses em uma grande variedade delas, além de aumentos de exposição proporcionais à dose a partir de 60 mg (ou 1 mg/kg).
Absorção
Após dose subcutânea do Denosumabe (substância ativa), a biodisponibilidade foi de 61% e as concentrações séricas máximas (Cmáx), de 6 mcg/mL (faixa de 1 a 17 mcg/mL), ocorreram em 10 dias (faixa de 2 a 28 dias). Em seguida à Cmáx os níveis séricos diminuíram, sendo a meia-vida de 26 dias (faixa de 6 a 52 dias) durante o período de 3 meses (faixa de 1,5 a 4,5 meses). Cinquenta e três por cento dos pacientes não apresentaram quantidades mensuráveis do Denosumabe (substância ativa) 6 meses pós-dose.
Distribuição
Nem acúmulo nem alteração da farmacocinética do Denosumabe (substância ativa) foram observados com o passar do tempo após doses múltiplas de 60 mg por via subcutânea 1 vez a cada 6 meses.
Metabolismo
O Denosumabe (substância ativa) é composto unicamente de aminoácidos e carboidratos, como imunoglobulina nativa. Com base em dados não clínicos, espera-se que o metabolismo do Denosumabe (substância ativa) siga as vias de eliminação da imunoglobulina, resultando em degradação para pequenos peptídeos e aminoácidos individualizados.
Eliminação
O Denosumabe (substância ativa) é composto unicamente de aminoácidos e carboidratos, como imunoglobulina nativa, e não se prevê que sejam eliminados por meio de mecanismos metabólicos hepáticos (como enzimas do citocromo P450, ou CYP). Tomando-se por base os dados não clínicos, prevê-se que a eliminação do Denosumabe (substância ativa) seguirá as vias de eliminação da imunoglobulina, resultando em degradação para pequenos peptídeos e aminoácidos individualizados.
Interações medicamentosas
Em um estudo com 17 mulheres na fase pós-menopausa com osteoporose, foi administrado midazolam (oral, 2 mg) duas semanas após uma dose única de Denosumabe (substância ativa) (subcutâneo, 60 mg), que corresponde ao tempo máximo de efeitos farmacodinâmicos de Denosumabe (substância ativa). O Denosumabe (substância ativa) não afetou a farmacocinética de midazolam que é metabolizado pelo citocromo P450 3A4 (CYP3A4). Isto indica que Denosumabe (substância ativa) não altera a farmacocinética de medicamentos metabolizados pelo CYP3A4.
Idosos (a partir de 65 anos de idade)
A idade não foi considerada um fator significativo na farmacocinética do Denosumabe (substância ativa) em uma análise farmacocinética da população de pacientes de 28 a 87 anos.
Crianças e adolescentes (até 18 anos de idade)
Não há dados farmacocinéticos disponíveis sobre pacientes pediátricos.
Raça
A farmacocinética do Denosumabe (substância ativa) não foi afetada pelo fator raça em mulheres na fase de pós-menopausa nem em pacientes com câncer de mama sob tratamento de ablação hormonal.
Insuficiência renal
Em um estudo com 55 pacientes que apresentavam graus variados de função renal, incluindo-se os que se submetiam a diálise, o grau de insuficiência renal não teve nenhum efeito sobre a farmacocinética nem sobre a farmacodinâmica do Denosumabe (substância ativa). Portanto, não é necessário ajuste de dose para pacientes com insuficiência renal.
Insuficiência hepática
Não foram conduzidos estudos clínicos para avaliar o efeito da insuficiência hepática sobre a farmacocinética do Denosumabe (substância ativa).
Cuidados de Armazenamento
O produto deve ser armazenado sob refrigeração (2ºC a 8ºC). Proteger da luz. Não o congelar.
A seringa deve ser mantida em sua embalagem original até o momento de uso do produto.
Número do lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o na embalagem original.
Todo medicamento deve ser mantido fora do alcance das crianças.
Aspecto físico/características organolépticas
Solução transparente, incolor à ligeiramente amarelada, que pode conter resíduos de partículas proteináceas translúcidas ou brancas.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
VENDA SOB PRESCRIÇÃO MÉDICA
MS: 1.0244.0008
Farm. Resp.: Monica Carolina Dantas Pedrazzi - CRF-SP 30.103
Importado por:
Amgen Biotecnologia do Brasil Ltda. Rua Patrícia Lúcia de Souza, 146. Taboão da Serra – SP
CNPJ: 18.774.815/0001-93
Fabricado por:
Amgen Manufacturing Limited Juncos - Porto Rico
® Marca Registrada
SAC 0800 264 0800
[email protected]