Comparamos o preço de Blincyto 38Mcg 1 Ampola 10Ml, veja o menor preço

R$ 11.765,00
BBiológicos
14
ofertasMelhores preços a partir de R$ 11.765,00 até R$ 14.917,00
Menor preço
vendido por Mundial Farma
economize
21.13%
R$ 11.765,00
vendido por Maranata Medicamentos
economize
15.29%
R$ 12.635,97
vendido por Onco Express Medicamentos Especiais e Oncológicos
economize
11.81%
R$ 13.156,00
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
vendido por Saúde Farma Medicamentos
economize
9.50%
R$ 13.500,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
vendido por Life Medicamentos
economize
9.32%
R$ 13.526,70
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Farma Visa
economize
9.32%
R$ 13.526,70
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Imune Farma Medicamentos Especiais
economize
9.32%
R$ 13.526,80
vendido por Farma Ame
economize
9.32%
R$ 13.526,85
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Farma Silva
economize
9.31%
R$ 13.528,00
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por OncoExpresso Medicamentos
economize
9.23%
R$ 13.540,00
vendido por Fast Medicamentos
economize
5.52%
R$ 14.093,60
vendido por Drogaria Dinâmica
economize
5.52%
R$ 14.094,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Facilita Medicamentos
economize
5.48%
R$ 14.100,00
vendido por Oncolog Medicamentos Especiais
R$ 14.917,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Para que serve
Blinatumomabe é o ingrediente ativo de Blincyto.
Blincyto é usado para tratar adultos com leucemia linfoblástica aguda. A leucemia linfoblástica aguda é um câncer das células brancas (glóbulos brancos) do sangue no qual um tipo específico de glóbulo branco cresce descontroladamente.
Como Blincyto funciona?
Este medicamento funciona ativando o sistema imunológico para atacar e destruir esses glóbulos brancos cancerígenos anormais.
Contraindicação
Não use Blincyto
- Se você for alérgico (hipersensível) a blinatumomabe ou a qualquer dos ingredientes de Blincyto;
- Se você não tiver certeza, converse com seu médico ou farmacêutico antes de receber Blincyto.
Como usar
Siga as instruções de seu médico, enfermeiro ou farmacêutico ao receber Blincyto. Se tiver quaisquer outras perguntas em relação à infusão de Blincyto, entre em contato com seu médico, enfermeiro ou farmacêutico.
Como Blincyto é administrado
A administração de Blincyto será feita por meio de uma veia (via intravenosa) continuamente por 4 semanas por uma bomba de infusão, seguida de uma pausa de 2 semanas durante a qual você não receberá a infusão (este é o ciclo de tratamento 1). Após essa pausa de 2 semanas, seu médico determinará se você receberá ciclos adicionais.
O número de ciclos de tratamento e a dose que você receberá dependerão de sua tolerabilidade e da resposta ao tratamento com Blincyto; seu médico discutirá com você por quanto tempo você receberá Blincyto. Além disso, o tratamento poderá ser interrompido, a depender de sua tolerabilidade a Blincyto.
Recomenda-se hospitalização no mínimo pelos primeiros 9 dias do primeiro ciclo e pelos primeiros 2 dias do segundo ciclo. Para todos os inícios e reinícios de ciclos subsequentes (por exemplo, se o tratamento for interrompido por 4 horas ou mais), recomenda-se supervisão por um profissional de saúde ou hospitalização.
Instruções detalhadas para seu médico, enfermeiro e farmacêutico sobre como preparar e infundir Blincyto estão incluídas na bula destinada ao profissional da saúde. Seu médico irá determinar quando sua bolsa de infusão de Blincyto será trocada, o que poderá variar desde uma troca diária até uma troca a cada 4 dias. A taxa de infusão pode ser mais rápida ou mais lenta a depender de quantas vezes a bolsa será trocada.
Seu primeiro ciclo
Para a primeira semana de infusão de Blincyto, a bomba será ajustada para infundir uma dose de 9 microgramas ao dia. Após o julgamento de seu médico, sua dose de Blincyto aumentará para 28 microgramas ao dia para as semanas de tratamento 2, 3 e 4.
Você pode não ser capaz de notar a diferença entre as bolsas de infusão de 9 microgramas ao dia e de 28 microgramas ao dia.
Os ciclos seguintes
Se seu médico determinar que você deve receber mais ciclos de Blincyto, sua bomba será ajustada para infundir uma dose de 28 microgramas ao dia.
Medicamentos administrados antes de cada ciclo de Blincyto
Antes que Blincyto lhe seja administrado, você receberá outros medicamentos (pré-medicação) para ajudar a reduzir as reações à infusão e outros efeitos colaterais possíveis. Estes podem incluir corticosteroides.
Cateter de infusão
Se você tiver um cateter para infusão, é muito importante manter a área em torno do cateter limpa; caso contrário você poderá ter uma infecção. Seu médico ou enfermeiro irá lhe mostrar como cuidar do local do cateter.
Bomba de infusão e administração intravenosa
Não ajuste as configurações da bomba, mesmo se houver um problema com a bomba ou com os sons de alarme da bomba. Quaisquer alterações na função da bomba podem resultar em doses muito altas ou muito baixas.
Entre em contato com seu médico ou enfermeiro imediatamente
- Se houver algum problema com a bomba ou com os sons de alarme da bomba;
- Se a bolsa de infusão esvaziar antes da programação de troca de bolsa;
- Se a bomba de infusão parar inesperadamente.
As atividades diárias normais não afetarão a forma como a bomba de infusão e a administração intravenosa funcionam. Entre em contato com o seu médico ou enfermeiro se tiver perguntas sobre suas atividades diárias e a função da bomba.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento de seu médico.
O que devo fazer quando eu me esquecer de usar Blincyto?
Em caso de dúvidas, procure orientação do farmacêutico ou do seu médico.
Precauções
Informe ao seu médico se qualquer dos casos seguintes for aplicável Blincyto pode não ser adequado
- Se você teve previamente problemas neurológicos (por exemplo, convulsões, perda de memória, confusão, desorientação, perda de equilíbrio ou dificuldade para falar);
- Se você tiver uma infecção ativa;
- Se você teve previamente uma reação pós-infusão ao usar Blincyto.
Informe ao seu médico ou farmacêutico imediatamente se você experimentar qualquer dos seguintes sintomas enquanto estiver recebendo Blincyto, visto que pode ser necessário tratar a reação e ajustar sua dose
- Se você apresentar convulsões, dificuldade para falar ou fala arrastada, confusão e desorientação ou perda de equilíbrio;
- Se você desenvolver calafrios ou tremores ou se sentir quente; você deve medir sua temperatura para verificar a ocorrência de febre – estes podem ser sintomas de uma infecção;
- Se você desenvolver uma reação a Blincyto em qualquer momento durante a infusão, que pode incluir tontura, inchaço da face, dificuldade para respirar, chiado no peito ou erupções cutâneas;
- Se você apresentar dor de estômago grave e persistente, com ou sem náusea e vômito, visto que estes podem ser sintomas de uma condição grave e potencialmente fatal conhecida como pancreatite (inflamação do pâncreas).
Seu médico ou enfermeiro irá monitorar os sinais e sintomas dessas reações.
Outros medicamentos e Blincyto
Informe ao seu médico ou enfermeiro se estiver tomando ou se tomou recentemente ou se puder vir a tomar quaisquer outros medicamentos, incluindo medicamentos obtidos sem prescrição médica.
Informe ao seu médico ou enfermeiro se você acha que pode precisar de vacinações em um futuro próximo, incluindo as necessárias para viajar para outros países. Algumas vacinas não podem ser administradas 2 semanas antes, no mesmo período ou alguns meses após você receber o tratamento com Blincyto. O seu médico verificará se você precisa de vacinação.
Gravidez e amamentação
Os efeitos de Blincyto em mulheres grávidas não são conhecidos.
Informe ao seu médico ou enfermeiro se você estiver grávida, se achar que está grávida ou se você pretender engravidar. Seu médico ajudará você a pesar os riscos e os benefícios em relação ao uso de Blincyto durante a gravidez.
Mulheres que podem engravidar devem usar anticoncepcionais durante o tratamento. Você também deve fazê-lo por 48 horas após o último tratamento. Converse com o seu médico ou enfermeiro sobre métodos contraceptivos adequados.
Informe ao seu médico ou enfermeiro caso você engravide durante o tratamento com Blincyto. O seu médico pode precisar conversar com você sobre precauções ao usar vacinação para o seu bebê.
Não é conhecido se os ingredientes no Blincyto passam para o leite materno. Informe ao seu médico ou enfermeiro se estiver amamentando ou planejando amamentar.
Você não deve amamentar durante o tratamento com Blincyto e por pelo menos 48 horas após o último tratamento.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Dirigir e operar máquinas
Não dirija, não opere máquinas pesadas e não se envolva em atividades perigosas enquanto estiver recebendo Blincyto. Blincyto pode ocasionar problemas neurológicos como tonturas, convulsões e confusão.
Informe ao seu médico se você está fazendo uso de algum outro medicamento.
Não use este medicamento sem o conhecimento de seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como todos os medicamentos, Blincyto pode causar certos efeitos colaterais, embora nem todos os pacientes os experimentem. Alguns desses efeitos colaterais podem ser sérios.
Informe ao seu médico imediatamente se você apresentar qualquer dos seguintes ou uma combinação dos seguintes efeitos colaterais
- Calafrios, tremores, febre, aumento da frequência cardíaca, redução da pressão arterial, dor muscular, fadiga, tosse, dificuldade para respirar, confusão, vermelhidão, inchaço ou hematoma na área afetada ou no local da linha de infusão – estes podem ser sinais de uma infecção;
- Eventos neurológicos: agitação (ou tremor), confusão, distúrbios da função cerebral (encefalopatia), dificuldade em se comunicar (afasia), convulsões;
- Febre, inchaço, calafrios, redução ou aumento da pressão arterial e líquido nos pulmões, que podem se tornar graves – estes podem ser sinais da síndrome de liberação de citocinas;
- Se você apresentar dor de estômago grave e persistente, com ou sem náusea e vômito, visto que estes podem ser sintomas de uma condição grave e potencialmente fatal conhecida como pancreatite (inflamação do pâncreas).
O tratamento com Blincyto pode causar uma redução das contagens de certos glóbulos brancos, com ou sem febre (neutropenia febril ou neutropenia) ou pode levar ao aumento das concentrações sanguíneas de potássio, ácido úrico e fosfato e redução das concentrações sanguíneas de cálcio (síndrome de lise tumoral). O seu médico conduzirá exames de sangue regulares durante o tratamento com Blincyto.
Outros efeitos colaterais incluem
Reações muito comuns (ocorrem em mais de 10% dos pacientes que utilizam este medicamento)
- Infecções no sangue incluindo por bactérias, fungos, vírus ou outros tipos de infecção;
- Redução das contagens de certos glóbulos brancos, com ou sem febre, neutropenia (febril), redução das contagens de glóbulos vermelhos, redução das contagens de plaquetas;
- Febre, inchaço, calafrios, redução ou aumento da pressão arterial e líquido nos pulmões, que podem se tornar graves (síndrome de liberação de citocinas);
- Não ser capaz de dormir;
- Dor de cabeça
- Aumento da frequência cardíaca (taquicardia);
- Pressão baixa;
- Tosse;
- Erupção cutânea;
- Dor nas costas, dor óssea;
- Febre (pirexia), inchaço nas mãos, tornozelos ou pés;
- Aumento das concentrações de enzimas hepáticas (ALT, AST, GGT).
Reações comuns (ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento)
- Ativação excessiva de glóbulos brancos associada a inflamação;
- Aumento das contagens de glóbulos brancos no sangue, redução das contagens de certos glóbulos brancos (linfopenia), doença dos linfonodos;
- Reação alérgica;
- Complicações ocorridas após tratamento de câncer que levam ao aumento das concentrações sanguíneas de potássio, ácido úrico e fosfato e redução das concentrações sanguíneas de cálcio (síndrome de lise tumoral);
- Confusão, desorientação;
- Distúrbios da função cerebral (encefalopatia) como dificuldade em se comunicar (afasia), formigamento da pele (parestesia), convulsões, dificuldade de raciocínio ou processamento de pensamentos, dificuldades de memória;
- Agitação (ou tremor), sonolência, dormência, tontura;
- Pressão arterial alta (hipertensão);
- Rubor;
- Chiado no peito ou dificuldade para respirar (dispneia), tosse com fleuma;
- Aumento da bilirrubina no sangue;
- Dor nas extremidades;
- Calafrios, dor torácica ou outra dor;
- Baixas concentrações de anticorpos chamados “imunoglobulinas”, que ajudam o sistema imunológico a combater infecções (diminuição das imunoglobulinas);
- Altas concentrações de algumas enzimas, incluindo enzimas sanguíneas e hepáticas;
- Aumento de peso;
- Sobredosagem, incluindo por acidente.
Reações incomuns (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento)
- Febre, inchaço, calafrios, redução ou aumento da pressão arterial e do líquido nos pulmões, que podem ser graves e que podem ser fatais (tempestade de citocinas);
- Dificuldade para falar;
- Problemas em nervos que afetam cabeça e pescoço, como distúrbios visuais, dificuldade em movimentos faciais, dificuldade em ouvir e dificuldade de deglutição (distúrbios dos nervos cranianos).
Informe ao seu médico, dentista ou farmacêutico se você notar qualquer reação adversa. Informe também à empresa pelo SAC.
Atenção: Este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe ao seu médico. Informe também à empresa através do seu serviço de atendimento.
Composição
Cada frasco-ampola contém:
|
12,5 mcg/mL* | |
| Blinatumomabe |
38,5 mcg |
|
Excipientes** |
q.s. |
*Após a reconstituição com 3 mL de Água Estéril para Injetáveis sem conservantes, o volume total resultante de solução reconstituída é de 3,1 mL e cada mL contém 12,5 mcg de blinatumomabe.
**Mono-hidrato de ácido cítrico, di-hidrato de trealose, cloridrato de lisina, polissorbato 80, hidróxido de sódio.
Cada frasco de Solução estabilizante IV contém:
|
10 mL | |
|
Excipientes* |
q.s. |
*Mono-hidrato de ácido cítrico, cloridrato de lisina, polissorbato 80, hidróxido de sódio, água para injetáveis.
Superdosagem
Entre em contato com o seu médico ou enfermeiro imediatamente se
- Houver um problema com a bomba ou com os sons de alarme de sua bomba;
- Você achar que pode ter recebido mais Blincyto do que deveria, por exemplo, a bolsa de infusão esvaziar antes do prazo planejado para a troca da bolsa.
Em caso de utilização de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou a bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Os resultados de um teste in vitro nos hepatócitos de seres humanos sugerem que blinatumomabe não afetou a atividade das enzimas do CYP450.
A elevação transitória das citocinas pode afetar a atividade das enzimas do CYP450. Com base nos modelos farmacocinéticos baseados fisiologicamente (PBPK), o efeito da elevação transitória das citocinas sobre a atividade das enzimas do CYP450 é inferior a 30%, com duração inferior a uma semana; o efeito sobre as exposições a substratos CYP450 sensíveis são menores que 2 vezes. Assim, a elevação das citocinas mediada por blinatumomabe parece ter um baixo potencial de interação medicamentosa clinicamente significativa.
Ação da Substância
No Estudo 1, a segurança e a eficácia deste medicamento em comparação com o standard of care (SOC) foram avaliadas em um estudo randomizado, aberto, multicêntrico. Os pacientes elegíveis tinham ? 18 anos de idade com LLA de linhagem B recidivada ou refratária (tinham > 5% de blastos na medula óssea e alguma recidiva em qualquer momento após transplante alogênico de células-tronco hematopoiéticas (TCTH-alo), primeira recidiva não tratada com a duração da primeira remissão < 12 meses ou refratária após a última terapia).
Os pacientes foram randomizados em uma razão 2:1 para receber este medicamento ou 1 de 4 regimes de quimioterapia padrão pré-especificada (SOC), selecionada pelo investigador. A randomização foi estratificada por idade (< 35 anos versus. ? 35 anos de idade), terapia de resgate anterior (sim versus. não) e TCTH-alo anterior (sim versus. não), conforme avaliado no momento do consentimento. Os dados demográficos e características basais foram bem equilibrados entre os dois braços (vide Tabela 1).
Tabela 1. Dados Demográficos e Características Basais no Estudo 1
|
Característica |
Este medicamento (N = 271) |
Quimioterapia SOCa (N = 134) |
|
Idade | ||
|
Mediana, anos (mín., máx.) |
37 (18, 80) |
37 (18, 78) |
|
Média, anos (DP) |
40,8 (17,1) |
41,1 (17,3) |
|
< 35 anos, n (%) |
123 (45,4) |
60 (44,8) |
|
? 35 anos, n (%) |
148 (54,6) |
74 (55,2) |
|
? 65 anos, n (%) |
33 (12,2) |
15 (11,2) |
|
? 75 anos, n (%) |
10 (3,7) |
2 (1,5) |
|
Homens, n (%) |
162 (59,8) |
77 (57,5) |
|
Raça, n (%) | ||
|
Índios Americanos ou Nativos do Alasca |
4 (1,5) |
1 (0,7) |
|
Asiáticos |
19 (7,0) |
9 (6,7) |
|
Negros (ou Afro-Americanos) |
5 (1,8) |
3 (2,2) |
|
Múltiplos |
2 (0,7) |
0 |
|
Nativos do Havaí ou Outra Ilha do Pacífico |
1 (0,4) |
1 (0,7) |
|
Outros |
12 (4,4) |
8 (6,0) |
|
Brancos |
228 (84,1) |
112 (83,6) |
|
Terapia de resgate anterior |
164 (60,5) |
80 (59,7) |
|
TCTH-alo anteriorb |
94 (34,7)
|
46 (34,3) |
|
Status no Grupo Cooperativo do Leste - n (%) |
|
|
|
0 |
96 (35,4) |
52 (38,8) |
|
1 |
134 (49,4) |
61 (45,5) |
|
2 |
41 (15,1) |
20 (14,9) |
|
Desconhecido |
0 |
1 (0,7) |
|
Refratários após tratamento de resgate - n (%) |
|
|
|
Sim |
87 (32,1) |
34 (25,4) |
|
Não |
182 (67,2) |
99 (73,9) |
|
Desconhecido |
2 (0,7) |
1 (0,7) |
|
Máximo de blastos na medula óssea central/local - n (%) |
|
|
|
? 5% |
0 |
0 |
|
> 5 a < 10% |
9 (3,3) |
7 (5,2) |
|
10 a < 50% |
60 (22,1) |
23 (17,2) |
|
? 50% |
201 (74,2) |
104 (77,6) |
|
Desconhecido |
1 (0,4) |
0 |
a SOC = standard of care.
b TCTH-alo = transplante alogênico de células-tronco hematopoiéticas.
Este medicamento foi administrado como uma infusão intravenosa contínua. No primeiro ciclo, a dose inicial foi de 9 mcg/dia para a semana 1, em seguida 28 mcg/dia nas 3 semanas restantes. A dose-alvo de 28 mcg/dia foi administrada no ciclo 2 e nos ciclos subsequentes iniciando no dia 1 de cada ciclo. O ajuste de dose foi possível em caso de eventos adversos. Dos 267 pacientes que receberam este medicamento, o número mediano de ciclos de tratamento foi 2 (variação: 0 a 9 ciclos); dos 109 pacientes que receberam quimioterapia padrão (SOC), o número mediano de ciclos de tratamento foi 1 (variação: 1 a 4 ciclos).
O desfecho primário foi sobrevida global (SG). O estudo demonstrou melhora estatisticamente significativa na SG para os pacientes tratados com este medicamento em comparação com a quimioterapia padrão (SOC). Em pacientes com 0 terapias de resgate anteriores, a razão de risco para SG foi 0,60 (0,39, 0,91), em pacientes com 1 terapia de resgate anterior, a razão de risco para SG foi 0,59 (0,38, 0,91) e em pacientes com mais de 2 terapias de resgate anteriores, a razão de risco para SG foi 1,13 (0,64, 1,99). O benefício de SG foi independente de transplante; resultados consistentes foram observados após recenseamento no momento do TCTH. Ver Figura 1 e Tabela 2 para os resultados de eficácia do Estudo 1.
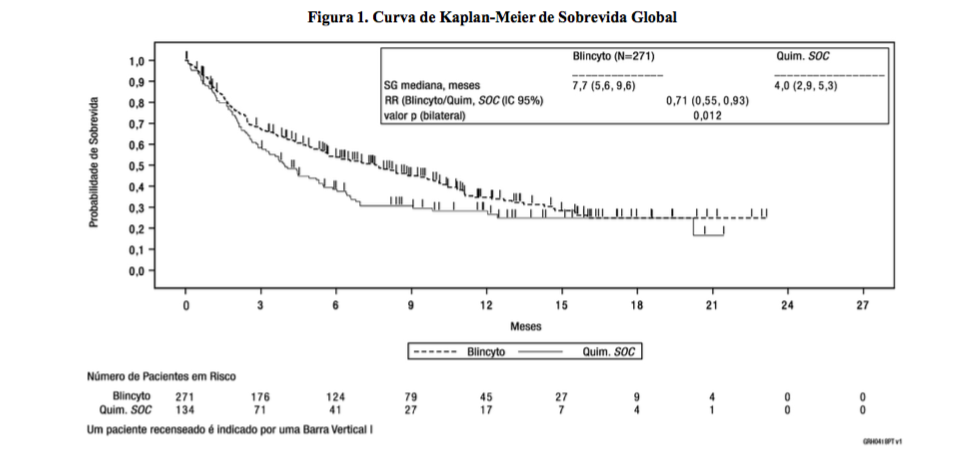
Tabela 2. Resultados de eficácia em pacientes ? 18 anos de idade com Leucemia Linfoblástica Aguda (LLA) de linhagem B recidivada ou refratária com cromossomo Philadelphia negativo (Estudo 1)
|
Este medicamento (N = 271) |
Quimioterapia SOC (N = 134) | |
|
Sobrevida global | ||
|
Mediana, meses [IC 95%] |
7,7 (5,6, 9,6) |
4,0 (2,9 a 5,3) |
|
Relação de Risco [IC 95%]a |
0,71 (0,55, 0,93) | |
|
Valor pb |
0,012 | |
|
Remissão Completa (RC) | ||
|
RCc/RCh*d/RCie, n (%) [IC 95%] |
119 (43,9) (37,9, 50,0) |
33 (24,6) (17,6, 32,8) |
|
Diferença no tratamento [IC 95%] |
19,3 (9,9, 28,7) | |
|
Valor pb |
< 0,001 | |
|
RC, n (%) [IC 95%] |
91 (33,6) (28,0, 39,5) |
21 (15,7) (10,0, 23,0) |
|
Diferença no tratamento [IC 95%] |
17,9 (9,6, 26,2) | |
|
Valor pf |
< 0,001 | |
|
Duração da RC/RCh*/RCig | ||
|
Mediana, meses [IC 95%] |
7,3 (5,8, 9,9) |
4,6 (1,8, 19) |
|
Sobrevida livre de Eventosh | ||
|
Estimativa em 6 meses% [IC 95%] |
30,7 (25, 36,5) |
12,5 (7,2, 19,2) |
|
Razão de Risco [IC 95%] |
0,55 (0,43, 0,71) | |
|
Resposta DRMj para RC/RCh*/RCi | ||
|
n1/n2 (%)j [IC 95%] |
74/97 (76,3) (66,6, 84,3) |
16/33 (48,5) (30,8, 66,5) |
a. Com base no modelo de Cox estratificado.
b. O valor p foi derivado a partir do teste de log-rank estratificado.
c. A RC foi definida como ? 5% de blastos na medula óssea, sem evidência de doença e recuperação completa das contagens sanguíneas periféricas (plaquetas > 100.000/microlitros e contagem absoluta de neutrófilos [CAN] > 1.000/microlitros).
d. A RCh* (remissão completa com recuperação hematológica parcial) foi definida como ? 5% de blastos na medula óssea, sem evidência de doença e recuperação parcial das contagens sanguíneas periféricas (plaquetas > 50.000/microlitros e CAN > 500/microlitros).
e. RCi (remissão completa com recuperação hematológica incompleta) foi definida como ? 5% de blastos na medula óssea, sem evidência de doença e recuperação incompleta das contagens sanguíneas periféricas (plaquetas > 100.000/microlitros ou CAN > 1.000/microlitros).
f. O valor p foi derivado por meio de teste de Cochran-Mantel-Haenszel.
g. A duração da RC/RCh*/RCi foi definida como o tempo desde a primeira resposta à recidiva ou morte, o que ocorrer primeiro. A recidiva foi definida como recidiva hematológica (blastos na medula óssea > 5% após RC) ou uma recidiva extramedular.
h. O tempo de SLE foi calculado a partir da randomização até a data de avaliação da doença indicando uma recidiva após alcançar uma RC/RCh*/RCi ou morte, o que ocorrer primeiro. Os pacientes que não alcançarem uma RC/RCh*/RCi dentro de 12 semanas do início do tratamento são considerados falhas de tratamento e recebem uma duração da SLE de 1 dia.
i. Resposta DRM (doença residual mínima) foi definida como DRM por PCR ou citometria de fluxo < 1 x 10-4.
j. n1: número de pacientes que alcançaram resposta DRM e RC/RCh*/RCi; n2: número de pacientes que alcançaram RC/RCh*/RCi.
No Estudo 2, a segurança e a eficácia deste medicamento foram avaliadas em um estudo em regime aberto, multicêntrico, de braço único. Os pacientes elegíveis tinham ? 18 anos de idade com LLA de linhagem B recidivada ou refratária com cromossomo Philadelphia negativo (recidivada com a duração da primeira remissão ? 12 meses no primeiro resgate ou recidivada ou refratária após a primeira terapia de resgate ou recidivada dentro de 12 meses do TCTH alogênico, e tinham ? 10% de blastos na medula óssea).
Este medicamento foi administrado como uma infusão intravenosa contínua. No primeiro ciclo, a dose inicial foi de 9 mcg/dia para a semana 1, em seguida 28 mcg/dia nas 3 semanas restantes. A dose-alvo de 28 mcg/dia foi administrada no ciclo 2 e nos ciclos subsequentes iniciando no dia 1 de cada ciclo. O ajuste de dose foi possível em caso de eventos adversos. A população tratada incluiu 189 pacientes que receberam pelo menos uma infusão deste medicamento; o número mediano de ciclos de tratamento foi dois (variação: 1 a 5). Os pacientes que responderam a este medicamento, mas recidivaram posteriormente tiveram a opção de ser tratados novamente com este medicamento. Entre os pacientes tratados, a idade mediana era de 39 anos (variação: 18 a 79 anos), 64 dos 189 (33,9%) haviam sido submetidos a um TCTH antes de receber este medicamento e 32 dos 189 (16,9%) haviam recebido mais de 2 terapias de resgate anteriores.
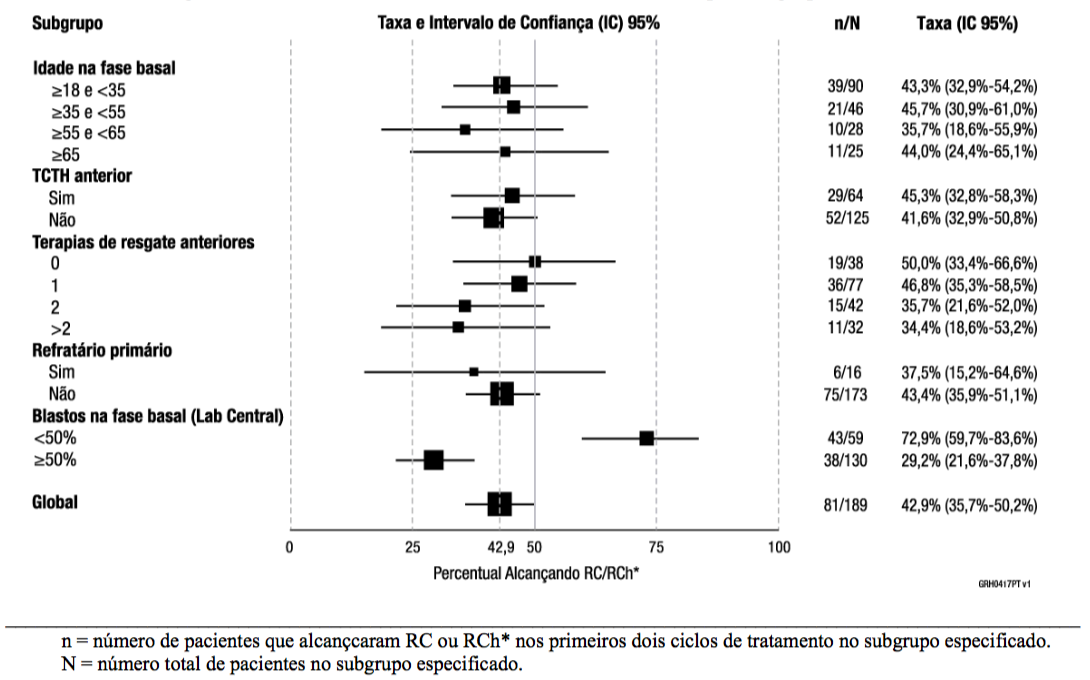
O desfecho primário foi a taxa de RC/RCh* dentro de dois ciclos de tratamento com este medicamento. Oitenta e um dos 189 (42,9%) pacientes alcançaram RC/RCh* dentro dos primeiros dois ciclos de tratamento com a maioria das respostas (64 de 81) ocorrendo dentro do ciclo 1 de tratamento. Ver Tabela 3 e Figura 2 para os resultados de eficácia do Estudo 2. Quatro pacientes alcançaram RC durante os ciclos subsequentes, resultando em uma taxa de RC cumulativa de 35,4% (67 de 189; IC 95%: 28,6% a 42,7%). Trinta e dois dos 189 (16,9%) pacientes foram submetidos a um TCTH alogênico em RC/RCh* induzida com este medicamento.
Tabela 3. Resultados de eficácia em pacientes ? 18 anos de idade com Leucemia Linfoblástica Aguda (LLA) de linhagem B recidivada ou refratária com cromossomo Philadelphia negativo (Estudo 2).
|
N = 189 | |
|
Remissão completa (RC)a/Remissão completa com recuperação hematológica parcial (RCh*)b, n (%) [IC 95%] |
81 (42,9%) [35,7% a 50,2%] |
|
RC, n (%) [IC 95%] |
63 (33,3%) [26,7% a 40,5%] |
|
RCh*, n (%) [IC 95%] |
18 (9,5%) [5,7% a 14,6%] |
|
Medula óssea hipoplásica ou aplásica sem blastoc, n (%) [IC 95%] |
17 (9%) [5,3% a 14,0%] |
|
Remissão parciald, n (%) [IC 95%] |
5 (2,6%) [0,9% a 6,1%] |
|
Sobrevida livre de recidivae (SLR) mediana para RC/RCh* [IC 95%] |
5,9 meses [4,8 a 8,3 meses] |
|
Sobrevida global mediana [IC 95%] |
6,1 meses [4,2 a 7,5 meses] |
a. A RC foi definida como ? 5% de blastos na medula óssea, sem evidência de doença, e recuperação completa das contagens sanguíneas periféricas (plaquetas > 100.000/microlitros e contagem absoluta de neutrófilos [CAN] > 1.000/microlitros).
b. A RCh* foi definida como ? 5% de blastos na medula óssea, sem evidência de doença, e recuperação parcial das contagens sanguíneas periféricas (plaquetas > 50.000/microlitros e CAN > 500/microlitros).
c. Medula óssea hipoplásica ou aplásica sem blastos foi definida como ? 5% de blastos na medula óssea, sem evidência de doença, recuperação insuficiente das contagens periféricas: plaquetas ? 50.000/microlitros e/ou CAN ? 500/microlitros.
d. A remissão parcial foi definida como 6% a 25% de blastos na medula óssea com uma redução de pelo menos 50% em relação à fase basal.
e. A recidiva foi definida como recidiva hematológica (blastos na medula óssea > 5% após RC) ou uma recidiva extramedular.
Figura 2. Taxa de RC/RCh* Durante os Primeiros Dois Ciclos por Subgrupo (Estudo 2)
Para avaliar melhor a sobrevida, foi conduzida uma análise com ponto de referência pré-especificado comparando respondedores e não respondedores na semana 5 dos ciclos 1 e 2. A sobrevida global mediana foi de 11,2 meses (IC 95%: 7,8 meses para não estimável) entre os pacientes que alcançaram RC/RCh* (N = 60) e 3,0 meses (IC 95%: 2,4 a 4 meses) entre não respondedores (N = 101) na análise do ciclo 1. A sobrevida global mediana foi de 9,9 meses (IC 95%: 6,8 meses para não estimável) entre os indivíduos que alcançaram RC/RCh* (N = 79), e 2,7 meses (IC 95%: 1,6 a 4,5 meses) entre os não respondedores (N = 50) na análise do ciclo 2.
Em uma análise exploratória pré-especificada, 60 dos 73 pacientes avaliáveis com DRM com RC/RCh* (82,2%) também tinham uma resposta DRM (definida como DRM por PCR < 1 x 10-4).
No Estudo 3, a segurança e a eficácia deste medicamento foram avaliadas em um estudo aberto, multicêntrico, de escalonamento de dose em 36 pacientes (incluindo 23 pacientes tratados em uma dose equivalente à dose proposta no registro) ? 18 anos de idade com LLA de linhagem B recidivada e/ou refratária (primeira ou maior recidiva, refratária, ou recidiva após transplante de células-tronco hematopoiéticas [TCTH]). Quinze dos 36 (41,7%) pacientes haviam passado por transplante de células-tronco hematopoiéticas (TCTH) alogênico antes de receber este medicamento. A taxa de remissão completa/remissão completa com recuperação hematológica parcial (RC/RCh*) foi de 69,4% [25 dos 36 pacientes (IC 95%: 51,9% a 83,7%): 15 (41,7%; IC 95%: 25,5% a 59,2%) RC; 10 (27,8%; IC 95%: 14,2% a 45,2%) RCh*]. Vinte e dois dos 25 (88%) pacientes com RC hematológica também tiveram respostas Doença Residual Mínima (DRM) (definida como DRM por PCR < 1 x 10-4). A duração mediana da remissão foi de 8,9 meses, a sobrevida livre de recidiva (SLR) mediana foi de 7,6 meses. A sobrevida global (SG) mediana foi de 9,8 meses.
No Estudo 4, este medicamento foi avaliado em um estudo de confirmação, multicêntrico, de braço único em 116 pacientes ? 18 anos de idade com DRM incluindo 5 pacientes (4,3%) com LLA Philadelphia positivo. O desfecho primário foi a proporção de pacientes que alcançaram resposta DRM completa definida pela ausência de DRM após um ciclo de tratamento com este medicamento. A percentagem de pacientes que alcançaram resposta DRM completa após um ciclo de tratamento foi de 77,9% (IC 95%: 69,1% a 85,1%).
Uma análise agrupada de dados multicêntricos e históricos (Estudo 5) em pacientes adultos com LLA recidivada ou refratária (n = 694 com dados de RC disponíveis; n = 1.112 com dados de SG disponíveis) foi realizada para fornecer um resumo dos principais resultados clínicos entre pacientes que receberam terapia de resgate. A definição da RC incluiu pacientes que experimentaram recuperação completa da medula óssea com recuperação completa da contagem sanguínea periférica bem como alguns pacientes em alguns centros com recuperação parcial da contagem sanguínea periférica. Os dados históricos incluíram pacientes com LLA recidivada ou refratária que tiveram recaída dentro de 12 meses do tratamento inicial, foram refratários a tratamento(s) anterior(s), recaíram dentro de 12 meses de um TCTH alogênico, ou estavam em um segundo ou posterior tratamento de resgate. Entre esses pacientes, a taxa de RC, ajustada ao perfil do paciente no Estudo 2 foi de 23,8% (IC 95%: 19,8% a 27,5%) e a SG mediana foi de 3,3 meses (IC 95%: 2,8 a 3,6 meses).
Características Farmacológicas
Farmacodinâmica
Foram observadas respostas farmacodinâmicas imunes consistentes nos pacientes estudados. Durante a infusão intravenosa contínua ao longo de 4 semanas, a resposta farmacodinâmica foi caracterizada pela ativação e redistribuição inicial das células T, rápida depleção das células B periféricas, e elevação transitória das citocinas.
A redistribuição das células T periféricas (ou seja, adesão das células T ao endotélio dos vasos sanguíneos e/ou a transmigração para os tecidos) ocorreu após o início da infusão deste medicamento ou do escalonamento de dose. As contagens de células T inicialmente declinaram dentro de 1 a 2 dias e em seguida voltaram aos valores basais dentro 7 a 14 dias na maioria dos pacientes. O aumento das contagens de células T acima da fase basal (expansão das células T) foi observado em poucos pacientes.
As contagens de células B periféricas diminuíram rapidamente a valores indetectáveis durante o tratamento nas doses ? 5 mcg/m2/dia ou ? 9 mcg/dia na maioria dos indivíduos. Não foi observada recuperação da contagem de células B periféricas durante o período de 2 semanas sem tratamento com este medicamento entre os ciclos de tratamento. Ocorreu depleção incompleta das células B nas doses de 0,5 mcg/m2/dia e 1,5 mcg/m2/dia e em poucos não respondedores em doses mais altas.
As citocinas incluindo IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-?, e IFN-? foram medidas, e IL-6, IL-10, e IFN-? estavam mais elevadas. A elevação transitória das citocinas foi observada nos primeiros 2 dias após o início da infusão deste medicamento. As concentrações elevadas de citocinas voltaram ao valor basal dentro de 24 a 48 horas durante a infusão. Nos ciclos de tratamento subsequentes, a elevação das citocinas ocorreu em menos pacientes com menor intensidade em comparação com as 48 horas iniciais do primeiro ciclo de tratamento.
Classe farmacológica
Anticorpo bispecífico ativador das células T (BiTE).
Mecanismo de ação
Blinatumomabe é um anticorpo bispecífico ativador das células T (BiTE) que se liga especificamente ao CD19 expresso na superfície das células de origem da linhagem B e ao CD3 expresso na superfície das células T. Ele ativa as células T endógenas conectando o CD3 no complexo do receptor de células T (TCR) com o CD19 nas células B benignas e malignas. A atividade antitumoral da imunoterapia com blinatumomabe não é dependente das células T contendo um TCR específico ou de antígenos peptídicos apresentados pelas células cancerosas, mas é de natureza policlonal e independente das moléculas do antígeno leucocitário humano (HLA) das células-alvo. Blinatumomabe medeia a formação de uma sinapse citolítica entre a célula T e a célula tumoral, liberando enzimas proteolíticas para matar tanto as células-alvo em proliferação quanto as que estão em repouso. Blinatumomabe é associado com regulação positiva transitória das moléculas de adesão celular, produção de proteínas citolíticas, liberação de citocinas inflamatórias, e proliferação de células T, e resulta na eliminação de células CD19+.
Imunogenicidade
Tal como acontece com todas as proteínas terapêuticas, há potencial para imunogenicidade. A imunogenicidade deste medicamento foi avaliada usando um imunoensaio de rastreamento com tecnologia de detecção eletroquimioluminescence (ECL) para a detecção da ligação de anticorpos antiblinatumomabe. Para pacientes cujos soros foram positivos no teste de imunoensaio, foi realizado um ensaio biológico in vitro para detectar anticorpos neutralizantes.
Nos estudos clínicos de pacientes adultos com LLA tratados com este medicamento, menos de 2% tiveram teste positivo para anticorpos antiblinatumomabe. Dos pacientes que desenvolveram anticorpos antiblinatumomabe, a maioria teve atividade neutralizante in vitro.
A formação de anticorpos antiblinatumomabe pode afetar a farmacocinética deste medicamento.
Se houver suspeita de formação dos anticorpos antiblinatumomabe com um efeito clinicamente significativo, o Serviço de Atendimento ao Consumidor (SAC) da Amgen deverá ser contactado.
A detecção da formação de anticorpos antiblinatumomabe é altamente dependente da sensibilidade e especificidade do ensaio. Adicionalmente, a incidência observada de formação de anticorpos (incluindo anticorpo neutralizante) em um ensaio pode ser influenciada por vários fatores, incluindo a metodologia do ensaio, a manipulação da amostra, o tempo de coleta da amostra, medicamentos concomitantes, e doença subjacente. Por essas razões, a comparação da incidência de anticorpos antiblinatumomabe com a incidência de anticorpos a outros produtos pode não ser correta.
Propriedades farmacocinéticas
A farmacocinética de blinatumomabe parece linear ao longo de uma variação de dose de 5 a 90 mcg/m2/dia (aproximadamente equivalente a 9 a 162 mcg/dia) em pacientes adultos. Após infusão intravenosa contínua, a concentração sérica no estado de equilíbrio dinâmico (Css) foi alcançada dentro de um dia e permaneceu estável ao longo do tempo. O aumento nos valores médios de Css foi aproximadamente proporcional à dose na variação testada. Nas doses clínicas de 9 mcg/dia e 28 mcg/dia para o tratamento de LLA recidivada/refratária, a Css média (DP) foi de 211 (258) pg/mL e 621 (502) pg/mL, respectivamente.
Distribuição
O volume de distribuição médio estimado baseado na fase terminal (Vz) foi de 4,52 (2,89) L com infusão intravenosa contínua de blinatumomabe.
Metabolismo
A via metabólica de blinatumomabe não foi caracterizada. Tal como outras terapias com proteínas, espera-se que este medicamento seja degradado em pequenos peptídeos e aminoácidos pelas vias catabólicas.
Eliminação
O clearance sistêmico médio estimado com infusão intravenosa contínua em pacientes recebendo blinatumomabe nos estudos clínicos foi de 2,92 (2,83) L/h. A meia vida média foi de 2,11 (1,42) horas. Quantidades insignificantes de blinatumomabe foram excretadas na urina nas doses clínicas testadas.
Peso corpóreo, Área de superfície corpórea, Sexo, e Idade
Uma análise farmacocinética da população foi realizada para avaliar os efeitos das características demográficas sobre a farmacocinética de blinatumomabe. Os resultados sugerem que idade (18 a 80 anos de idade), sexo, peso corpóreo (44 a 134 kg), e área de superfície corpórea (1,39 a 2,57 m2) não influenciam a farmacocinética de blinatumomabe.
Comprometimento renal
Não foram conduzidos estudos formais de farmacocinética de blinatumomabe em pacientes com comprometimento renal.
A análise farmacocinética mostrou uma diferença de aproximadamente 2 vezes nos valores médios de clearance de blinatumomabe entre indivíduos com disfunção renal moderada e função renal normal. Visto que foi observada elevada variabilidade intraindividual (CV% até 95,6%), e os valores de clearance em indivíduos com comprometimento renal ficaram essencialmente dentro da variação observada em indivíduos com função renal normal, não é esperado impacto da função renal clinicamente significativo na evolução clínica.
Dados de Segurança Pré-Clínica/Toxicologia não clínica
Blinatumomabe apresenta reação cruzada apenas no chimpanzé. Consequentemente, os dados de segurança pré-clínica com blinatumomabe são limitados. Uma molécula substituta murina com atividade biológica semelhante em camundongos foi desenvolvida para uso em testes não clínicos. Dados de estudos de duração de até 13 semanas com a molécula substituta murina em camundongos revelaram os efeitos farmacológicos esperados incluindo liberação de citocinas, redução nas contagens de leucócitos, depleção de células B, reduções nas células T, e redução da celularidade em tecidos linfoides. Estas alterações foram revertidas após a cessação do tratamento.
Mutagenicidade
Não foram conduzidos estudos de mutagenicidade com blinatumomabe; contudo, não é esperado que blinatumomabe altere o DNA ou os cromossomos.
Carcinogenicidade
Não foram conduzidos estudos de carcinogenicidade com blinatumomabe.
Cuidados de Armazenamento
Caso a bolsa de administração intravenosa seja entregue em sua casa, as bolsas intravenosas contendo a solução de Blincyto para infusão chegarão em uma embalagem especial. Não abra a embalagem. Conserve a embalagem em temperatura ambiente. Não refrigere ou congele a embalagem.
A embalagem será aberta por seu enfermeiro e as bolsas intravenosas serão armazenadas sob refrigeração (2°C a 8°C) até a infusão, e a infusão será feita em não mais que 10 dias após a preparação. Uma vez à temperatura ambiente (até 27°C) a solução será infundida dentro de 96 horas.
Não descarte quaisquer medicamentos pelo esgoto ou no lixo doméstico. Pergunte ao seu médico, enfermeiro ou farmacêutico como descartar os medicamentos que você não usa mais.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use o medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS 1.0244.0011.
Farm. Resp.:
Monica Carolina Dantas Pedrazzi
CRF-SP 30.103.
Importado por:
Amgen Biotecnologia do Brasil
Rua Patrícia Lucia de Souza, 146 – Jd. das Oliveiras.
Taboão da Serra – SP.
CNPJ: 18.774.815/0001-93.
Fabricado por:
Boehringer Ingelheim Pharma GmbH & Co. KG
Biberach - Alemanha.
Embalado por:
Amgen Manufacturing Limited
Juncos - Porto Rico.
Venda sob prescrição médica.