Comparamos o preço de Gilenya 0,5Mg Cápsulas C 28, veja o menor preço

R$ 8.540,00
RReferência
16
ofertasMelhores preços a partir de R$ 8.540,00 até R$ 12.959,59
Menor preço
vendido por OncoExpresso Medicamentos
economize
34.10%
R$ 8.540,00
vendido por Farma Visa
economize
26.02%
R$ 9.587,70
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Imune Farma Medicamentos Especiais
economize
26.02%
R$ 9.587,80
vendido por Life Medicamentos
economize
20.92%
R$ 10.248,70
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Farma Ame
economize
20.92%
R$ 10.248,85
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Agille Speciality
economize
20.74%
R$ 10.271,52
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Grupo Hera Medicamentos
economize
20.33%
R$ 10.325,00
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Farma Silva
economize
18.54%
R$ 10.557,50
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Agille Medicamentos
economize
18.29%
R$ 10.589,20
Preço Válido para compra em Boleto
vendido por Onco Express Medicamentos Especiais e Oncológicos
economize
16.74%
R$ 10.790,00
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
vendido por Drogaria Dinâmica
economize
16.70%
R$ 10.794,92
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Facilita Medicamentos
economize
16.70%
R$ 10.795,97
vendido por Saúde Farma Medicamentos
economize
13.83%
R$ 11.167,80
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria!
vendido por Pague Menos
economize
10.49%
R$ 11.599,99
vendido por Fast Medicamentos
economize
8.92%
R$ 11.803,72
vendido por Oncolog Medicamentos Especiais
R$ 12.959,59
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
Para que serve
Gilenya é um medicamento sob prescrição médica tomado na forma de cápsulas, e é utilizado para tratar esclerose múltipla remitente recorrente.
Gilenya não cura a esclerose múltipla, mas ajuda a reduzir o número de recidivas que ocorrem e diminuir o acúmulo de problemas médicos devido à esclerose múltipla (progressão da doença).
Como Ginelya funciona?
Gilenya pode alterar a forma que o sistema imune do corpo funciona e ajuda a combater ataques ao sistema imune, afetando a habilidade de algumas células brancas sanguíneas de se moverem livremente no sangue e impedindo as células que causam inflamação de chegarem ao cérebro. Isto reduz os danos nos nervos causados pela esclerose múltipla.
Gilenya também pode ter um efeito direto e benéfico em certas células do cérebro (células neurais) envolvidas na reparação ou redução dos danos causados pela esclerose múltipla.
Em estudos clínicos, Gilenya demonstrou reduzir o número de ataques (por pouco mais da metade) e como uma consequência, reduziu o número de recidivas graves e recidivas que devem ser tratadas no hospital, prolongando o tempo sem recidivas e diminuindo a progressão da doença (por cerca de um terço).
Se você tiver perguntas sobre como Gilenya funciona ou porque este medicamento foi indicado para você, pergunte ao seu médico.
Contraindicação
Não tome Gilenya:
- Se você é alérgico (hipersensível) ao fingolimode ou a qualquer um dos excipientes de Gilenya listados na bula. Se você suspeitar ser alérgico, converse com seu médico sobre isso.
- Se você teve ataque do coração, angina instável, derrame ou suspeita de derrame, ou alguns tipos de insuficiência cardíaca nos últimos 6 meses.
- Se você teve certos tipos de batimentos irregulares ou anormais do coração (arriia), incluindo pacientes com um problema chamado QT prolongado visto no ECG, pressão sanguínea não controlada ou problemas graves de respiração durante o sono (apneia do sono grave) antes de iniciar o tratamento com Gilenya.
- Se você está tomando certos medicamentos que alteram o rio cardíaco.
- Se você tem problemas graves no fígado.
Como usar
Siga as instruções do seu médico cuidadosamente. Não exceda a dose recomendada.
Quanto Gilenya tomar
A dose é de uma cápsula ao dia (0,5 mg de fingolimode). ?
Como e quando tomar Gilenya
Tome Gilenya uma vez ao dia com meio copo de água. Gilenya pode ser tomado com ou sem alimentos.
Tomar Gilenya no mesmo horário todos os dias poderá ajudar você a se lembrar de quando tomar seu medicamento.
Por quanto tempo tomar Gilenya
Não pare de tomar Gilenya ou mude sua dose sem falar com seu médico.
Se você tiver dúvidas sobre por quanto tempo tomar Gilenya, fale com seu médico ou farmacêutico.
Se você parar de tomar Gilenya
Não pare de tomar Gilenya ou mude sua dose sem falar com seu médico. Gilenya ficará no seu corpo por até 2 meses após a interrupção. Sua contagem de células brancas sanguíneas (contagem linfocítica) também poderá ficar baixa durante este período de tempo e os efeitos descritos nesta bula ainda podem ocorrer.
Se você parar de tomar Gilenya por 1 dia ou mais durante seu primeiro mês de tratamento, ou se você parar mais que duas semanas após seu primeiro mês de tratamento com Gilenya, os efeitos iniciais na sua frenquência cardíaca podem ocorrer novamente. Quando você reiniciar o tratamento com Gilenya, seu médico pode decidir monitorar sua frequência cardíaca, pressão sanguínea a cada hora, realizar eletrocardiogramas ou manter você sob monitoramento durante a noite.
A dose máxima recomendada de Gilenya é de uma cápsula de 0,5 mg ao dia.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que devo fazer quando eu me esquecer de usar este medicamento?
Se você esquecer uma dose, tome a próxima dose conforme planejado. Não tome uma dose dobrada para compensar a dose esquecida.
Se você estiver tomando Gilenya há menos de 2 semanas e esqueceu de tomar a dose por um dia, entre em contato com seu médico imediatamente.
Seu médico pode decidir colocá-lo em observação no período até a próxima dose. Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Cuidados especiais ao tomar Gilenya:
- Será solicitado que você faça um ECG (eletrocardiograma) para verificar a saúde do seu coração antes que você inicie o tratamento com Gilenya e um segundo eletrocardiograma, ao final do período de observação de 6 horas após você tomar a primeira dose de Gilenya. Sua frequência cardíaca e pressão arterial também serão verificadas a cada hora, por um profissional de saúde, durante essas 6 horas do período de observação. No caso de um eletrocardiograma anormal ou diminuição da frequência cardíaca no final do período de observação de 6 horas, você pode ser observado por mais tempo e durante a noite, se necessário, por um profissional de saúde. A mesma recomendação pode se aplicar se você iniciar o tratamento novamente após uma pausa da terapia com Gilenya, dependendo de quanto tempo for a pausa do tratamento e em quanto tempo você está reiniciando o tratamento.
Verificar a condição de seu coração é particularmente importante se qualquer uma das seguintes condições se aplica a você. O seu médico pode decidir não utilizar Gilenya. Se o seu médico considerar que Gilenya é bom para você, ele (a) pode encaminhá-lo primeiro para um cardiologista (médico especializado em doenças do coração). Você também pode ser monitorado durante a noite, por um profissional de saúde, após tomar a primeira dose de Gilenya.
Informe seu médico antes de tomar Gilenya:
- Se você tiver a frequência cardíaca irregular ou anormal, uma doença grave no coração, pressão alta não controlada, histórico de derrame ou outra doença relacionada aos vasos sanguíneos no cérebro, se quando você dorme é gravemente afetado por incapacidade de respirar (apneia do sono não tratada) e se você apresenta risco ou possui distúrbios do rio cardíaco (chamado prolongamento QTc ou eletrocardiograma anormal dos batimentos cardíacos). Seu médico pode decidir não utilizar Gilenya se você apresenta ou já apresentou uma dessas condições.
- Se você estiver tomando medicamentos para batimentos cardíacos irregulares, tais como quinidina, procainamida, amiodarona ou sotalol.
- Se você sofre de frequência cardíaca baixa, se no início do tratamento com Gilenya você estiver tomando medicamentos que diminuem o rio cardíaco ou se você tiver histórico de perda súbita de consciência (desmaios). Seu médico pode decidir não utilizar Gilenya ou pode encaminhá-lo primeiro para um cardiologista para substituir por medicamentos que não diminuam rio cardíaco ou para decidir como você deve ser observado após tomar a primeira dose de Gilenya.
No início do tratamento, Gilenya pode causar a diminuição da frequência cardíaca. Gilenya também pode causar batimentos cardíacos irregulares principalmente após a primeira dose. Os batimentos cardíacos irregulares geralmente retornam ao normal em menos de um dia. A frequência cardíaca baixa geralmente retorna ao normal dentro de um mês. Se a sua frequência cardíaca diminuir após a primeira dose, você pode sentir tonturas, cansaço ou pode estar consciente do seu batimento cardíaco. Se a sua frequência cardíaca diminuir muito ou sua pressão arterial cair, você pode necessitar de tratamento imediato. Neste caso, você será monitorado durante a noite, por um profissional de saúde e o mesmo processo de observação ocorrido para sua primeira dose de Gilenya também será aplicável para a segunda dose.
Se alguma das condições abaixo se aplicar a você, informe seu médico antes de tomar Gilenya:
- Se você não tem nenhuma história de catapora ou não tenha sido vacinado contra o vírus varicela zoster. O seu médico vai testar a condição dos seus anticorpos contra esse vírus e vacinar você se não tiver anticorpos suficientes para este vírus. Neste caso, você vai começar o tratamento com Gilenya um mês após que o curso completo da vacinação for concluído.
- Se você tem uma resposta imune reduzida (devido a doenças ou medicamentos que suprimem o sistema imune). Você pode contrair infecções mais facilmente ou uma infecção que você já tem pode piorar. Gilenya diminui a contagem de células brancas sanguíneas (particularmente a contagem de linfócitos). As células brancas sanguíneas combatem infecções. Enquanto você estiver tomando Gilenya (e por até 2 meses após a descontinuação), você pode contrair infecções mais facilmente.
- Se você apresentar alguma infecção, fale com seu médico antes de tomar Gilenya. Qualquer infecção que você já tenha pode piorar. As infecções podem ser sérias e às vezes com riscos de vida. Antes de começar a tomar Gilenya , seu médico irá confirmar se você tem uma quantidade suficiente de células brancas no seu sangue. Durante seu tratamento com Gilenya , se você achar que está apresentando uma infecção, com sintomas de febre, ou como se tivesse com gripe, ou tiver dor de cabeça acompanhada por rigidez na nuca, sensibilidade a luz, náusea e ou confusão (estes podem ser sintomas de meningite), avise seu médico imediatamente. Se você acredita que sua esclerose múltipla está piorando (por exemplo, fraqueza ou alterações visuais) ou se você notar quaisquer sintomas novos ou pouco habituais, converse com o seu médico o mais breve possível, porque estes podem ser os sintomas de uma doença cerebral rara causada por uma infecção e chamada leucoencefalopatia multifocal progressiva (LMP).
- Se você planeja tomar uma vacina. Você não deve tomar alguns tipos de vacina (chamadas “vacinas com vírus vivos atenuados”) durante e até 2 meses após o tratamento com Gilenya.
- Se você tem ou teve distúrbios visuais ou outros sinais de inchaço na área da visão central posterior do olho (uma condição conhecida como edema macular), inflamação ou infecção do olho (uveíte) ou se você tem diabetes. Seu médico pode querer que você se submeta a um exame nos olhos antes de iniciar Gilenya e em intervalos regulares após o início do tratamento com Gilenya. A mácula é uma pequena área da retina posterior do olho que permite que você veja formatos, cores e detalhes clara e nitidamente (visão central). Gilenya pode causar o inchaço da mácula, uma condição conhecida como edema macular. O inchaço geralmente acontece durante os primeiros 4 meses de tratamento com Gilenya. Sua probabilidade de desenvolver edema macular é maior se você tiver diabetes ou se você teve uma inflamação no olho chamada uveíte. O edema macular pode causar alguns dos mesmos sintomas visuais que um ataque de esclerose múltipla (neurite ótica). Logo no início, pode não haver quaisquer sintomas. Certifique-se de informar seu médico sobre quaisquer alterações na sua visão. Seu médico pode querer que você se submeta a exames nos olhos, particularmente se o centro da sua visão ficar embaçado ou tiver sombras, se você desenvolver um ponto cego no centro da sua visão ou se você tiver problemas para enxergar cores ou detalhes.
- Se você tem problemas no fígado. Você precisará de um exame de sangue para verificar a sua função do fígado antes de começar a tomar Gilenya. Gilenya pode afetar sua função hepática. Você provavelmente não irá notar quaisquer sintomas, mas se você notar amarelamento da pele ou do branco dos olhos, urina escura ou náusea e vômito inexplicados durante o tratamento, avise seu médico imediatamente. Seu médico pode solicitar exames de sangue para verificar sua função hepática e pode considerar a descontinuação do tratamento se o problema no seu fígado for sério.
Informe seu médico imediatamente se você tiver algum dos seguintes sintomas ou doenças durante seu tratamento com Gilenya.
Uma condição denominada síndrome de encefalopatia posterior reversível foi raramente relatada em pacientes com esclerose múltipla tratados com Gilenya. Os sintomas podem incluir início repentino de dor de cabeça grave, confusão, convulsões e alterações visuais. Informe seu médico se você asentar prequalquer um desses sintomas durante seu tratamento com Gilenya. Um tipo de câncer denominado de carcinoma basocelular (CBC) foi reportado em pacientes com esclerose múltipla tratados com Gilenya. Converse com seu médico se você notar algum nódulo na pele (e.x. nódulos perolados brilhantes), manchas ou feridas abertas que não cicatrizam dentro de semanas (esses podem ser sinais de CBC).
Reações Adversas
Assim como todos os medicamentos, pacientes em tratamento com Gilenya podem apresentar efeitos adversos, embora nem todos os apresentem.
Alguns efeitos podem ocorrer com certa frequência, que está definida abaixo:
| Muito comum | Pode afetar mais de 1 em 10 pessoas |
| Comum | Pode afetar entre 1 e 10 a cada 100 pessoas |
| Incomum | Pode afetar 1 entre 10 a cada 1000 pessoas |
| Rara | Pode afetar 1 entre 10 a cada 10000 pessoas |
| Frequência desconhecida | Frequência não pode ser estimada através dos dados disponíves |
Comum:
- Bronquite com sintomas como tosse com catarro, dor no peito e febre.
- Gastroenterite com sintomas como vômitos, náuseas, diarreia e febre.
- Infecções por herpes zoster podem se apresentar com sintomas como bolhas, ardor, coceira ou dor na pele, tipicamente na parte superior do corpo ou no rosto. Outros sintomas podem ser febre e fraqueza, nos estágios iniciais da infecção, seguidas de entorpecimento coceira ou manchas vermelhas com dor intensa.
- Frequência cardíaca baixa (bradicardia).
- Um tipo de câncer de pele denominado carcinoma basocelular (CBC), que frequentemente se apresenta como nódulos perolados, embora também possa assumir outras formas.
Incomum:
- Pneumonia com sintomas como febre, tosse, dificuldade de respirar
- Edema macular (inchaço na área de visão central da retina posterior do olho) com sintomas como sombras e pontos cegos no centro da visão, visão borrada, dificuldade em ver cores e detalhes.
Raras:
- Uma condição denominada Síndrome de Encefalopatia Posterior Reversível. Os sintomas podem incluir início repentino de dor de cabeça grave, confusão, convulsões e alterações na visão.
Casos isolados:
- Irregularidade grave no batimento cardíaco, que é temporária e volta ao normal durante o período de observação de 6 horas.
Desconhecida:
- Reações alérgicas, incluindo sintomas de erupção cutânea ou coceira (urticária), inchaço dos lábios, língua ou face, que são mais prováveis de ocorrer no dia do início do tratamento com Gilenya.
- Doença cerebral rara causada por uma infecção e chamada leucoencefalopatia multifocal progressiva (LMP). Os sintomas de LMP podem ser semelhantes aos da esclerose múltipla (por exemplo, fraqueza ou alterações visuais).
- Infecções criptocócicas (um tipo de infecção fúngica), incluindo meningite criptocócica com sintomas de dor de cabeça acompanhados por rigidez da nuca, sensibilidade à luz, náusea e confusão.
Se você apresentar quaisquer destes sintomas, avise seu médico imediatamente.
Outros efeitos adversos
Muito comuns
- Infecção pelo vírus da gripe com sintomas como cansaço, calafrios, dor de garganta, dor nas articulações ou músculos, febre;
- Sensação de pressão ou dor nas bochechas e na testa (sinusite);
- Dor de cabeça;
- Diarreia;
- Dor nas costas;
- Exames de sangue mostrando níveis mais altos de enzimas do fígado;
- Tosse.
Se qualquer um destes sintomas afetar você gravemente, avise seu médico. ?
Comuns
- Micose, uma infecção fúngica da pele (tinea versicolor);
- Tontura;
- Formigamento ou dormência
- Dor de cabeça severa sempre acompanhada de náusea, vômitos e sensibilidade à luz (enxaqueca);
- Fraqueza;
- Coceira, vermelhidão, queimadura cutânea (eczema);
- Perda de cabelo;
- Coceira na pele;
- Perda de peso;
- Níveis aumentados de gordura (triglicérides) no sangue;
- Falta de ar;
- Resultados anormais no teste de função pulmonar após um mês de tratamento, mantendo-se estável depois disso e reversível após a descontinuação do tratamento;
- Depressão;
- Dor no olho;
- Visão borrada
- Hipertensão. Gilenya pode causar um leve aumento da pressão sanguínea;
- Nível baixo de células sanguíneas brancas (linfopenia e leucopenia).
Se qualquer um destes sintomas afetar você gravemente, avise seu médico.
Se você notar qualquer outro efeito adverso não mencionado nesta bula, por favor, informe seu médico ou farmacêutico.
População Especial
Idosos (acima de 65 anos de idade)
Experiências com Gilenya em idosos são limitadas. Converse com seu médico se você tiver quaisquer preocupações.
Crianças e adolescentes (abaixo de 18 anos de idade)
Gilenya não é indicado para uso em crianças e adolescentes uma vez que não foi estudado em pacientes com esclerose múltipla abaixo de 18 anos.
Gravidez e lactação
Você deve evitar engravidar enquanto estiver tomando Gilenya ou dentro de dois meses após a descontinuação, por causa do risco de prejudicar o feto. Fale com seu médico sobre os riscos associados, e sobre métodos confiáveis para o controle da gravidez que você deverá utilizar durante o tratamento e por 2 meses após a descontinuação do tratamento. Avise seu médico se você está grávida, acha que está grávida ou se está tentando engravidar. Se você engravidar enquanto estiver tomando Gilenya avise seu médico imediatamente. Você e seu médico irão decidir o que for melhor para você e para o bebê. Você não deverá amamentar enquanto estiver tomando Gilenya. Gilenya pode passar para o leite materno e existe um risco de efeitos adversos sérios para o lactente. Fale com seu médico antes de amamentar enquanto estiver tomando Gilenya. Consulte seu médico ou farmacêutico antes de tomar qualquer medicamento, se você estiver grávida ou amamentando.
Dirigir e operar máquinas
Seu médico dirá se sua doença permite que você dirija veículos ou opere máquinas com segurança. Não se espera que Gilenya afete na sua habilidade de dirigir ou operar máquinas. Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento. Não use este medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Composição
Cada cápsula de Gilenya contém:
0,56 mg de cloridrato de fingolimode, equivalente a 0,5 mg de fingolimode.
Excipientes:
Manitol, estearato de magnésio, óxido de ferro amarelo, dióxido de titânio, gelatina, tinta de impressão preta e tinta de impressão amarela. ?
Superdosagem
Se você tiver tomado muitas cápsulas de Gilenya de uma só vez ou se você tiver tomado a primeira dose de Gilenya por engano, entre em contato com seu médico imediatamente.
O seu médico pode decidir observá-lo, medindo a pressão arterial e frequência cardíaca a cada hora, realizar ECGs e ele pode decidir monitorá-lo durante a noite.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Interações farmacodinâmicas
Terapias antineoplásicas, imunomoduladoras ou imunossupressoras (incluindo corticosteroides) devem ser administradas concomitantemente com cautela devido ao risco de efeitos adicionais no sistema imune.
Decisões específicas como a dose e duração do tratamento concomitante com corticosteroides devem ser baseadas na avaliação clínica.
A coadministração de um curto período de corticosteroides (até 5 dias conforme protocolos de estudo) não aumentou a taxa global de infecção em pacientes tratados com fingolimode em estudos de fase III, comparando com o placebo.
Também deve-se ter cautela para introduzir Cloridrato de Fingolimode (substância ativa), quando os pacientes estiverem previamente em uso de terapias que possuem efeitos imune de longa duração, tais como natalizumabe, teriflunomida ou mitoxantrona.
Quando o fingolimode é usado com atenolol, há uma redução adicional de 15% na frequência cardíaca mediante a iniciação de fingolimode, um efeito não observado com diltiazem.
O tratamento com Cloridrato de Fingolimode (substância ativa) não deve ser iniciado em pacientes recebendo betabloqueadores, bloqueadores do canal de cálcio redutores da frequência cardíaca (como verapamil ou diltiazem), ou outras substâncias que podem diminuir a frequência cardíaca (por exemplo, ivabradina ou digoxina) devido aos potenciais efeitos aditivos na frequência cardíaca.
Se o tratamento com Cloridrato de Fingolimode (substância ativa) for considerado, deve-se procurar aconselhamento com um cardiologista a respeito da substituição por medicamentos que não reduzam a frequência cardíaca ou o monitoramento adequado durante o inicio do tratamento (deve durar pelo menos toda a noite).
Durante e até dois meses após o tratamento com Cloridrato de Fingolimode (substância ativa), a vacinação poderá ser menos eficaz.
O uso de vacinas vivas atenuadas pode apresentar risco de infecção e deve, portanto, ser evitado.
Interações farmacocinéticas
O fingolimode é eliminado principalmente via citocromo P450 4F2 (CYP4F2) e possivelmente por outras isoenzimas CYP4F.
Estudos em hepatócitos in vitro indicam que o CYP3A4 pode contribuir para a metabolização do fingolimode nos casos de forte indução do CYP3A4.
Potencial de fingolimode e fingolimode-fosfato para inibir o metabolismo de medicações concomitantes
Estudos de inibição in vitro utilizando microssomos agrupados do fígado humano e substratos metabólicos específicos de sonda demonstraram que fingolimode e fingolimode-fosfato possuem pouca ou nenhuma capacidade de inibir a atividade das enzimas CYP (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, ou CYP4A9/11 (apenas fingolimode)).
Portanto, é improvável que o fingolimode e o fingolimode-fosfato reduzam o clearance dos medicamentos que são eliminados principalmente através do metabolismo pelas principais isoenzimas CPY.
Potencial de fingolimode e fingolimode-fosfato de induzir seu próprio metabolismo e/ou o metabolismo de medicações concomitantes
O fingolimode foi examinado quanto a seu potencial de induzir a atividade de CYP3A4, CYP1A2, CYP4F2, e ABCB1 (P-gp) mRNA e CYP3A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, e CYP4F2 humanos em hepatócitos humanos primários.
O fingolimode não induziu o mRNA ou a atividade das diferentes enzimas CYP e ABCB1 a respeito do veículo controle. Portanto, nenhuma indução clinicamente relevante das enzimas CYP ou ABCB1 (P-gp) testadas pelo fingolimode é esperada em concentrações terapêuticas. Experimentos in vitro não forneceram indicações de indução do CYP pelo fingolimode-fosfato.
Potencial de fingolimode e fingolimode-fosfato de inibir o transporte ativo de medicações concomitantes
Com base em dados in vitro não se espera que o fingolimode ou que o fingolimode-fosfato, inibam a captação de medicações concomitantes e/ou biológicos transportados pelo o polipeptídio transportador de ânion orgânico 1B1 e 1B3 (OATP1B1, OATP1B3) ou o polipeptídio cotransportador de taurocolato de sódio (NTCP).
De maneira semelhante, não se espera que eles inibam o efluxo de medicações concomitantes e/ou biológicos transportados pela proteína resistente ao câncer de mama (BCRP), pela bomba de excreção do sal biliar (BSEP), pela proteína 2 associada à resistência a múltiplos medicamentos (MRP2) em concentrações terapêuticas.
Contraceptivos orais
A co-administração de fingolimode de 0,5 mg diariamente com contraceptivos orais (etinilestradiol e levonorgestrel) não provocou qualquer mudança na exposição à contraceptivos orais. A exposição a fingolimode e fosfato de fingolimode foram consistentes com os estudos anteriores.
Nenhum estudo de interação foi realizado com contraceptivos orais contendo outras progesteronas, entretanto, um efeito de fingolimode sobre a sua exposição não é esperado.
Ciclosporina
A farmacocinética do fingolimode de dose única não foi alterada durante a administração concomitante com ciclosporina em estado de equilíbrio, nem a farmacocinética de ciclosporina em estado de equilíbrio foi alterada pela administração de fingolimode de dose única, ou doses múltiplas (28 dias).
Esses dados indicam que é improvável que fingolimode reduza ou aumente o clearance de medicamentos eliminados principalmente pela CYP3A4. A inibição potente dos transportadores PgP, MRP2, e OATP1B1 não influencia a disposição de fingolimode.
Cetoconazol
A administração concomitante de cetoconazol 200 mg duas vezes ao dia em estado de equilíbrio e uma dose única de 5 mg de fingolimode levou a um aumento modesto na AUC de fingolimode e fingolimode-fosfato (aumento de 1,7 vez),pela inibição do CYP4F2.
Isoproterenol, atropina, atenolol, e diltiazem
A exposição à dose única de fingolimode e fingolimode-fosfato não foi alterada pela administração concomitante com isoproterenol ou atropina.
Da mesma forma, a farmacocinética de dose única de fingolimode e fingolimode-fosfato e a farmacocinética no estado de equilíbrio tanto do atenolol quanto do diltiazem não foram alteradas durante a administração concomitante dos últimos dois medicamentos com fingolimode.
Carbamazepina
A coadministração de carbamazepina 600 mg duas vezes ao dia no estado de equilíbrio e uma dose única de fingolimode 2 mg tiveram um fraco efeito na AUC do fingolimode e fingolimode-fosfato, diminuindo ambas em aproximadamente 40%. A relevância clínica dessa observação é desconhecida.
Análise farmacocinética da população para interações medicamentosas potenciais
Uma avaliação farmacocinética da população, realizada em pacientes com esclerose múltipla, não forneceu evidência para um efeito significativo da fluoxetina e da paroxetina (fortes inibidores do CYP2D6) sobre as concentrações de fingolimode ou fingolimode-fosfato.
Adicionalmente, as seguintes substâncias comumente prescritas não tiveram nenhum efeito clinicamente relevante (? 20%) sobre as concentrações de fingolimode ou fingolimode-fosfato: baclofeno, gabapentina, oxibutinina, amantadina, modafinil, amitriptilina, pregabalina, corticosteroides e anticoncepcionais orais.
Testes laboratoriais
Uma vez que o fingolimode reduz as contagens de linfócitos no sangue através da redistribuição em órgãos linfoides secundários, as contagens de linfócitos no sangue periférico não podem ser utilizadas para avaliar o status do subgrupo de linfócitos de um paciente tratado com Cloridrato de Fingolimode (substância ativa).
Testes laboratoriais que necessitam do uso de células mononucleares circulantes precisam de volumes maiores de sangue devido à redução no número de linfócitos circulantes.
Ação da Substância
Resultados de eficácia
A eficácia de Cloridrato de Fingolimode (substância ativa)(1) foi demonstrada em dois estudos que avaliaram doses diárias de 0,5 mg e 1,25 mg de Cloridrato de Fingolimode (substância ativa) em pacientes com esclerose múltipla remitente recorrente.
Os dois estudos incluíram pacientes que haviam apresentado pelo menos 2 recidivas clínicas durante os 2 anos antes da randomização ou pelo menos 1 recidiva clínica durante o 1° ano antes da randomização, e que haviam apresentado uma Escala de Estado de Incapacidade Expandida (EDSS) entre 0 e 5,5. Um terceiro estudo visando a mesma população de pacientes foi concluído após o registro de Cloridrato de Fingolimode (substância ativa).
Estudo D2301 (FREEDOMS)
O estudo D2301 (FREEDOMS) foi um estudo de fase III de 2 anos, randomizado, duplo-cego, placebo controlado em pacientes com esclerose múltipla recidiva-remitente que não haviam recebido betainterferona ou acetato de glatirâmer ao longo de pelo menos 3 meses anteriores e que não haviam recebido natalizumabe ao longo de pelo menos 6 meses anteriores.
Avaliações neurológicas foram realizadas na seleção, a cada 3 meses e no momento de suspeita da recidiva. Avaliações de MRI foram realizadas na seleção, mês 6, mês 12 e mês 24. O desfecho primário foi a taxa de recidiva anual.
A idade média era de 37 anos, a duração mediana da doença era de 6,7 anos e a pontuação mediana basal na EDSS foi de 2,0. Os pacientes foram randomizados para receber Cloridrato de Fingolimode (substância ativa) 0,5 mg (n = 425), Cloridrato de Fingolimode (substância ativa) 1,25 mg (n = 429) ou placebo (n = 418) por até 24 meses.
O tempo mediano recebendo o medicamento no estudo foi de 717 dias com 0,5 mg, 715 dias com 1,25 mg e 718,5 dias com placebo.
A taxa de recidiva anual foi significativamente menor em pacientes tratados com Cloridrato de Fingolimode (substância ativa) do que em pacientes que receberam placebo. O principal desfecho secundário foi o tempo até a progressão da incapacidade confirmada em 3 meses conforme medida por pelo menos um aumento de 1 ponto a partir do valor basal na EDSS (aumento de 0,5 ponto para pacientes com valor de basal de 5,5 na EDSS) mantido por 3 meses.
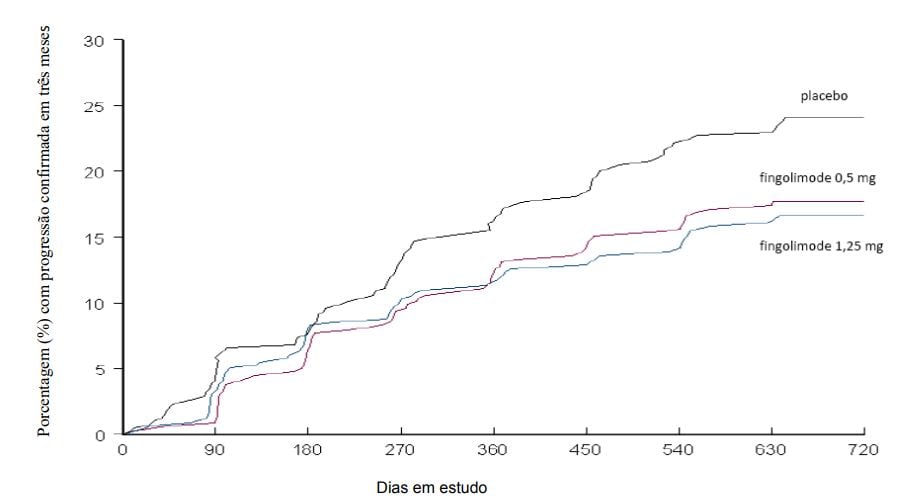
O tempo até o início da progressão da incapacidade confirmada em 3 meses foi significativamente retardado com o tratamento com Cloridrato de Fingolimode (substância ativa) em comparação com placebo. Não houve diferença significativa entre as doses de 0,5 mg e 1,25 mg em ambos os desfechos.
Os resultados para esse estudo estão demonstrados na Tabela 1 e Figuras 1 e 2.
Tabela 1 Resultados clínicos e de MRI do Estudo FREEDOMS
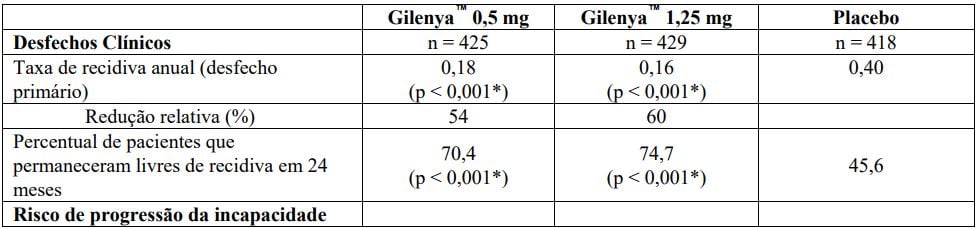
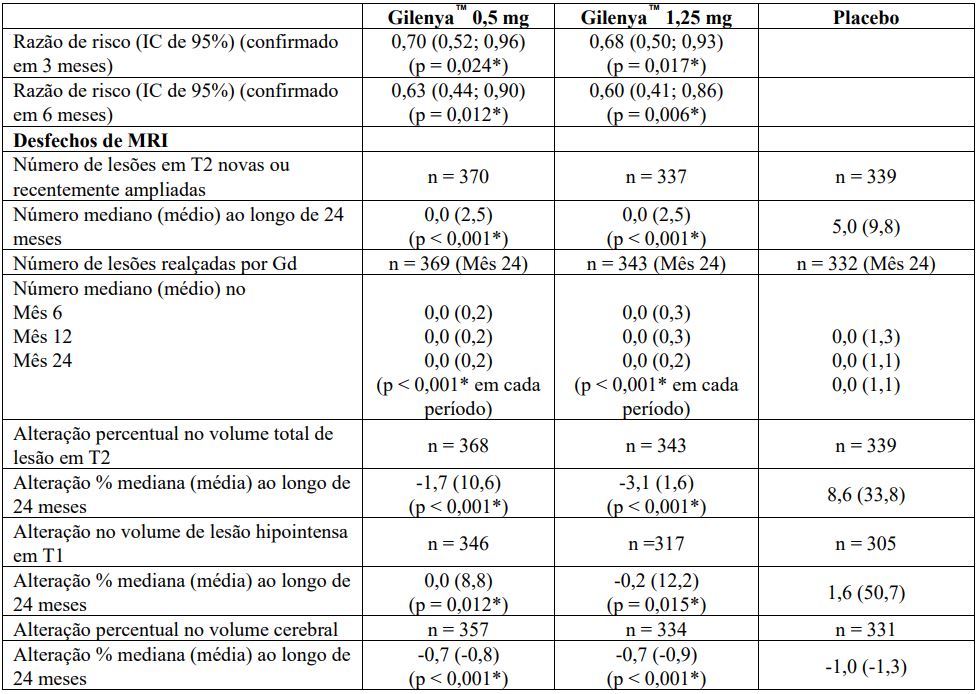
Todas as análises de desfechos clínicos foram intencionadas para tratar. As análises MRI usaram o conjunto de dados avaliável.
*Indica significância estatística versus placebo nível 0,05 bilateral.
Determinação de valores-p: ARR agregado por regressão binominal negativa ajustada por tratamento, país agrupado, número de recidivas nos últimos 2 anos e EDSS basal; percentual de pacientes mantendo regressão logística livre de recidiva ajustada por tratamento, país, número de recidiva nos últimos 2 anos, e EDSS basal; tempo até a progressão de deficiência confirmada em 3 meses/6 meses pelo modelo de riscos proporcionais de Cox ajustado por tratamento, país agrupado, EDSS basal, e idade; lesões em T2 novas/recentemente ampliadas por regressão binominal negativa ajustada por tratamento e país agrupado; lesões realçadas por Gd pelo rank ANCOVA ajustado por tratamento, país agrupado, e número basal das lesões realçadas por Gd; e alteração % na lesão e volume cerebral pelo rank ANCOVA ajustado por tratamento, país agrupado, e valor basal correspondente.
Figura 1 Gráfico de Kaplan-Meier do tempo até a primeira recidiva confirmada até o Mês 24 – Estudo FREEDOMS (população ITT)
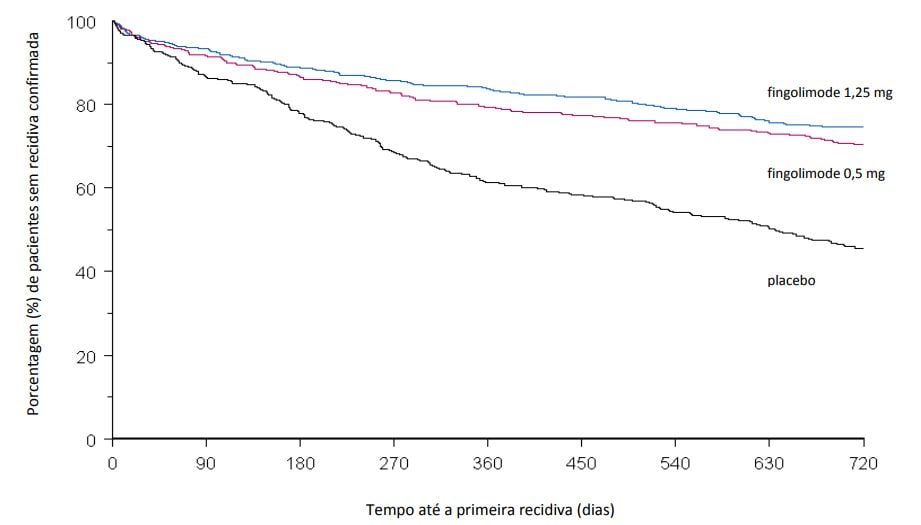
Figura 2 Gráfico cumulativo do tempo até a progressão da deficiência confirmada em 3 meses – Estudo FREEDOMS (população ITT)
Pacientes que completaram o estudo FREEDOMS (D2301) tinham a opção de entrar no estudo de extensão duplo-cego D2301E1(3). 920 pacientes do estudo principal entraram na extensão e foram todos tratados com fingolimode (n = 331 continuaram com 0,5 mg, 289 continuaram com 1,25 mg, 155 trocaram de placebo para 0,5 mg e 145 trocaram do placebo para 1,25 mg). 811 desses pacientes (88,2%) tiveram pelo menos 18 meses de acompanhamento na fase de extensão.
A duração máxima da exposição cumulativa a fingolimode 0,5 mg (principal + extensão) foi de 1.782 dias.
No mês 24 do estudo de extensão, os pacientes que receberam placebo no estudo principal tiveram reduções em taxa de recidiva anual de 55% após a mudança para fingolimode 0,5 mg (razão taxa de recidiva anual 0,45, 95% CI 0,32-0,62, p < 0,001). A taxa de recidiva anual para os pacientes que foram tratados com fingolimode 0,5 mg no estudo principal manteve-se baixa durante o estudo de extensão (taxa de recidiva anual de 0,10 no estudo de extensão).
Estudo D2309 (FREEDOMS II)
O estudo D2309 (FREEDOMS II) teve um desenho semelhante ao do estudo D2301 (FREEDOMS): o estudo foi de 2 anos, randomizado, duplo-cego, placebo-controlado, fase III, em pacientes com esclerose múltipla remitente recorrente da esclerose múltipla que não receberam qualquer betainterferona ou acetato de glatirâmer, pelo menos nos 3 meses anteriores e não receberam qualquer natalizumabe durante pelo menos os seis meses anteriores.
Foram realizadas avaliações neurológicas durante a triagem, a cada 3 meses, e no momento da suspeita de recidiva. Avaliações de MRI foram realizadas na triagem, mês 6, mês 12 e mês 24. O objetivo primário foi a taxa de recidiva anual.
A idade média foi 40,5 anos, a duração média da doença foi de 8,9 anos e a média da pontuação da EDSS no início do estudo foi de 2,5. Os pacientes foram randomizados para receber tratamento com Cloridrato de Fingolimode (substância ativa) 0,5 mg (n = 358) ou Cloridrato de Fingolimode (substância ativa) 1,25 mg (n = 370) ou placebo (n = 355) por até 24 meses.
O tempo médio com a medicação de estudo foi de 719 dias com 0,5 mg e 719 dias com placebo. Os pacientes randomizados para o braço com dose de fingolimode de 1,25 mg foram trocados de forma cega para receber fingolimode 0,5 mg quando os resultados do estudo de 2.301 ficaram disponíveis e confirmaram um melhor risco benefício da dose mais baixa.
A dose foi alterada para 113 pacientes (30,5%) nesse braço da dose, o tempo médio de fingolimode 1,25 mg neste braço foi de 496,1 dias e 209,8 dias para fingolimode 0,5 mg.
A taxa de recidiva anual foi significativamente inferior nos pacientes tratados com Cloridrato de Fingolimode (substância ativa) em relação aos pacientes que receberam placebo. O primeiro objetivo secundário chave foi a mudança no volume cerebral do valor basal. A perda de volume cerebral foi significativamente inferior no tratamento com Cloridrato de Fingolimode (substância ativa) em comparação com o placebo. O outro objetivo secundário chave foi a confirmação da progressão da incapacidade no tempo de três meses, medida por pelo menos aumento de 1 ponto do valor basal na EDSS (aumento de 0,5 ponto para pacientes com EDSS valor basal de 5,5) sustentado por 3 meses.
O risco de progressão de incapacidade para Cloridrato de Fingolimode (substância ativa) e placebo não foram estatisticamente diferentes.
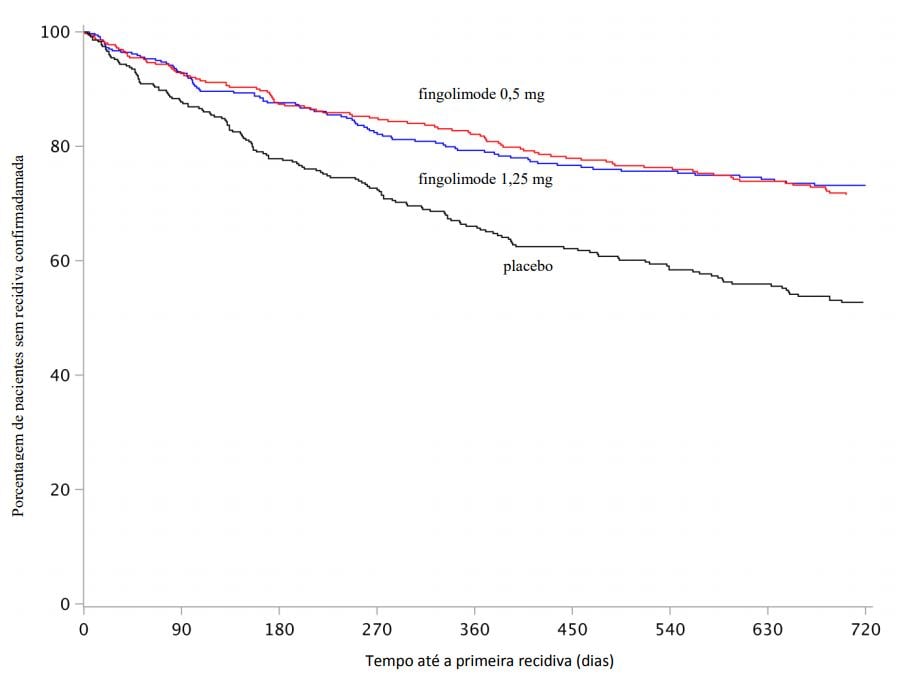
Não houve diferenças significativas entre as doses de 0,5 mg e 1,25 mg em qualquer um dos objetivos. Os resultados deste estudo são apresentados na Tabela 2 e na Figura 3.
Tabela 2 Resultados clínicos e de MRI do Estudo FREEDOMS II
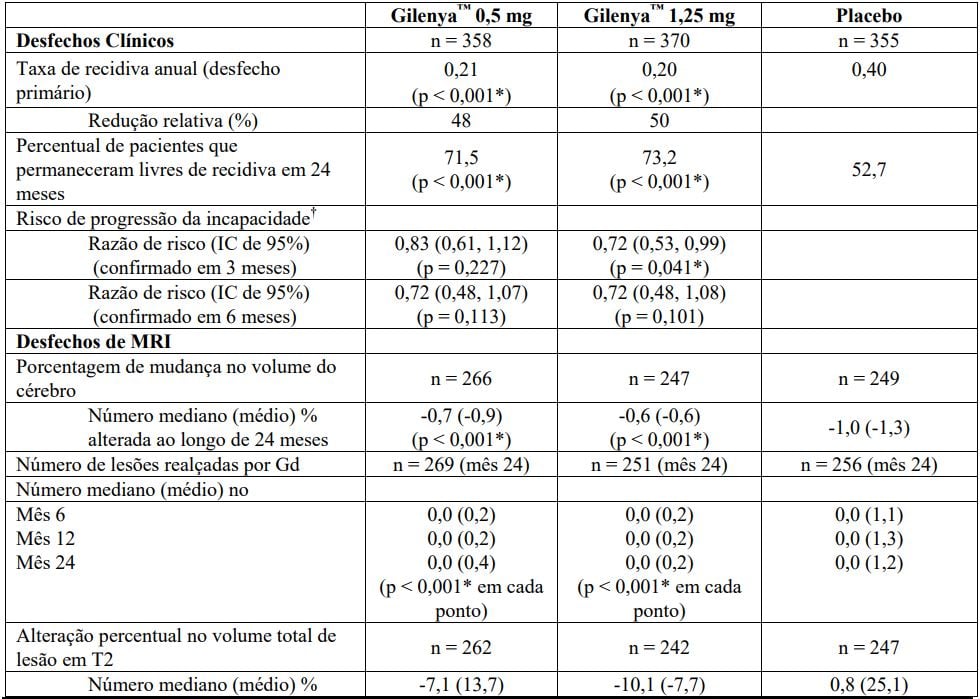
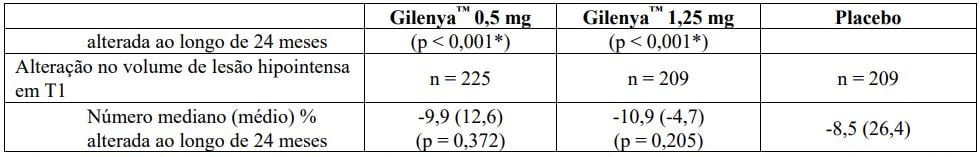
Todas as análises de desfechos clínicos foram intencionadas para tratar. As análises MRI usaram o conjunto de dados avaliável.
* Indica significância estatística versus placebo nível 0,05 bilateral.
Determinação de valores-p: ARR agregado por regressão binominal negativa ajustada por tratamento, país agrupado, número de recidivas nos últimos 2 anos e EDSS basal; percentual de pacientes mantendo regressão logística livre de recidiva ajustada por tratamento, país, número de recidiva nos últimos 2 anos, e EDSS basal; tempo até a progressão de deficiência confirmada em 3 meses/6 meses pelo modelo de riscos proporcionais de Cox ajustado por tratamento, país agrupado, EDSS basal, e idade; lesões em T2 novas/recentemente ampliadas por regressão binominal negativa ajustada por tratamento e país agrupado; lesões realçadas por Gd pelo rank ANCOVA ajustado por tratamento, país agrupado, e número basal das lesões realçadas por Gd; e alteração % na lesão e volume cerebral pelo rank ANCOVA ajustado por tratamento, país agrupado, e valor basal correspondente.
† Análises adicionais revelaram que resultados na população total não foram significativos devido às progressões falso positivas no subgrupo de pacientes com EDSS basal = 0 (n = 62, 8,7% da população do estudo). Em pacientes com EDSS > 0 (n = 651, 91,3% da população do estudo), fingolimode 0,5 mg demonstrou uma redução clinicamente relevante e estatisticamente significativa em relação ao placebo (HR = 0,70, IC (0,50, 0,98), p = 0,040), de acordo com estudo FREEDOMS.
Figura 3 Gráfico Kaplan-Meier para o tempo até a primeira recidiva confirmada até o Mês 24 – Estudo FREEDOMS II (população ITT)
Estudo D2302 (TRANSFORMS)
O Estudo D2302 (TRANSFORMS) foi um estudo de Fase III de 1 ano, randomizado, duplo-cego, duplo-mascarado, ativo-controlado (betainterferona 1a, 30 mcg, intramuscular, uma vez por semana) em pacientes com esclerose múltipla remitente recorrente que não haviam recebido natalizumabe nos últimos 6 meses.
A terapia anterior com betainterferona ou acetato de glatirâmer até o momento da randomização foi permitida.
Avaliações neurológicas foram realizadas na seleção, a cada 3 meses e no momento das suspeitas recidivas.
Avaliações por MRI foram realizadas na seleção e no mês 12. O desfecho primário foi a taxa de recidiva anual.
A idade média era de 36 anos, a duração mediana da doença era de 5,9 anos e a pontuação mediana na EDSS basal foi de 2,0.
Os pacientes foram randomizados para receber Cloridrato de Fingolimode (substância ativa) 0,5 mg (n = 431) ou Cloridrato de Fingolimode (substância ativa) 1,25 mg (n = 426) ou 30 microgramas de betainterferona 1a pela via intramuscular uma vez por semana (n = 435) por até 12 meses.
O tempo médio do estudo recebendo o medicamento de 365 dias com Cloridrato de Fingolimode (substância ativa) 0,5 mg, 354 dias com Cloridrato de Fingolimode (substância ativa) 1,25 mg e 361 dias com betainterferona 1a.
A taxa de recidiva anual foi significativamente menor em pacientes tratados com Cloridrato de Fingolimode (substância ativa) do que em pacientes que receberam betainterferona 1a IM.
Não houve diferença significativa entre as doses de 0,5 mg e 1,25 mg de Cloridrato de Fingolimode (substância ativa).
Os principais desfechos secundários foram o número de lesões em T2 novas ou recentemente ampliadas e o tempo até o início da progressão da deficiência confirmada em 3 meses conforme medida por pelo menos um aumento de 1 ponto a partir do valor de basal na EDSS (aumento de 0,5 ponto para aqueles com valor basal de 5,5 na EDSS) mantido por 3 meses.
O número de lesões em T2 novas ou recentemente ampliadas foi significativamente menor em pacientes tratados com Cloridrato de Fingolimode (substância ativa) do que em pacientes que receberam betainterferona 1a IM.
Não houve diferença significativa no tempo até a progressão de deficiência confirmada em 3 meses entre pacientes tratados com Cloridrato de Fingolimode (substância ativa) e betainterferona 1a IM em 1 ano.
Não houve diferença significativa entre as doses de 0,5 mg e 1,25 mg em quaisquer desfechos.
Os resultados para esse estudo são demonstrados na Tabela 3 e Figura 4.
Tabela 3 Resultados clínicos e de MRI do Estudo TRANSFORMS
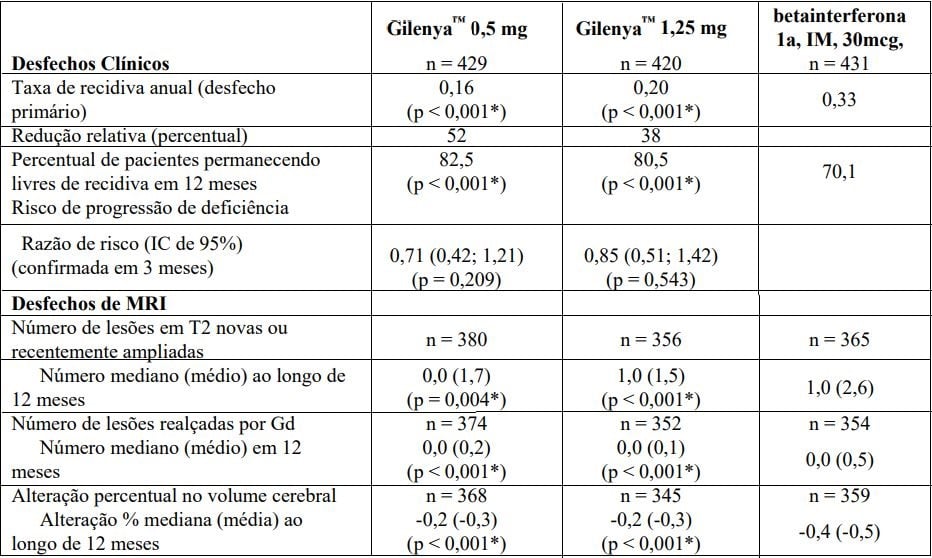
Todas as análises de desfechos clínicos foram intencionadas para tratar. As análises MRI usaram o conjunto de dados avaliável.
* Indica significância estatística versus betainterferona 1a IM no nível 0,05 bilateral.
Determinação de valores-p: ARR agregado por regressão binominal negativa ajustada por tratamento, país, número de recidivas nos últimos 2 anos e EDSS basal; porcentagem de pacientes mantendo regressão logística livre de recidiva ajustada por tratamento, país, número de recidiva nos últimos 2 anos, e EDSS basal; risco de progressão da deficiência pelo modelo de riscos proporcionais de Cox ajustado por tratamento, país, EDSS basal, e idade; lesões em T2 novas/recentemente ampliadas por regressão binominal negativa ajustada por tratamento, país, número de recidivas nos últimos 2 anos e EDSS basal; lesões realçadas por Gd pelo rank ANCOVA ajustado por tratamento, país, e número basal das lesões realçadas por Gd; e alteração % no volume cerebral pelo teste de soma de postos do rank Wilcoxon.
Figura 4 Gráfico Kaplan-Meier para o tempo até a primeira recidiva confirmada até o Mês 12 – Estudo TRANSFORMS (população ITT)
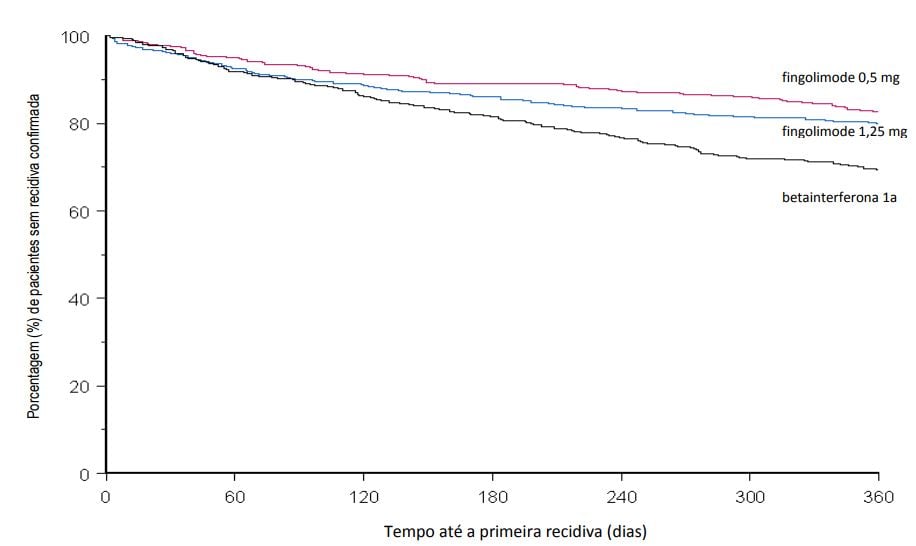
Os pacientes que completaram o estudo TRANSFORMS (D2302) tinham a opção de entrar na extensão de dose-cega. 1.030 pacientes do estudo principal entraram na extensão (estudo D2302E1), e foram tratados com fingolimode (n = 357 continuaram em 0,5 mg, 330 continuaram em 1,25 mg, 167 trocaram de betainterferona-1a para 0,5 mg e 176 trocaram de betainterferona-1a para 1,25 mg). 882 desses pacientes (85,9%) tiveram pelo menos 12 meses de acompanhamento na fase de extensão.
A duração máxima da exposição cumulativa a fingolimode 0,5 mg (estudo principal + extensão) foi de 1.594 dias.
No mês 12 de extensão do estudo, os pacientes que receberam betainterferona-1a i.m. no estudo principal tiveram reduções relativas na taxa de recidiva anual de 30% após mudar para o fingolimode 0,5 mg (taxa de recidiva anual = 0,70, p = 0,06).
A taxa de recidiva anual para os pacientes que foram tratados com fingolimode 0,5 mg no estudo principal foi baixa durante a combinação do estudo principal e da extensão (taxa de recidiva anual de 0,18 até ao mês 24).
Os resultados agrupados dos estudos D2301 (FREEDOMS) e D2302 (TRANSFORMS) demonstraram uma redução consistente da taxa na recidiva anual de Cloridrato de Fingolimode (substância ativa) em comparação com o comparador em subgrupos definidos por sexo, idade, terapia anterior para esclerose múltipla, atividade da doença ou níveis de deficiência basal.
Características farmacológicas
Código ATC: L04AA27.
Mecanismo de ação
O fingolimode é um modulador do receptor esfingosina-1-fosfato.
O fingolimode é metabolizado pela esfingosinaquinase ao metabólito ativo fingolimode-fosfato.
O fingolimode-fosfato se liga em concentrações nanomolares baixas aos receptores esfingosina-1-fosfato (S1P) 1, 3, e 4 localizados nos linfócitos, e cruza prontamente a barreira hematoencefálica para se ligar aos receptores S1P 1, 3, e 5 localizados nas células neurais no sistema nervoso central (SNC).
Agindo como um antagonista funcional de S1PR nos linfócitos, o fingolimode-fosfato bloqueia a capacidade dos linfócitos de egressar dos linfonodos, causando uma redistribuição, ao invés da depleção dos linfócitos.
Essa redistribuição reduz a infiltração de células linfocíticas, incluindo células pró-inflamatórias Th17, patogênicas no SNC, no qual elas seriam envolvidas em inflamação nervosa e dano de tecido nervoso.
Estudos em animais e experimentos in vitro indicam que o fingolimode pode também exercer efeitos benéficos na esclerose múltipla através da interação com receptores S1P em células neurais.
O fingolimode penetra no SNC, tanto em seres humanos como em animais, e demonstrou reduzir a astrogliose, desmielinização e perda neuronal. Além disso, o tratamento com fingolimode aumenta os níveis do fator neurotrópico derivado do cérebro (BDNF) no córtex, hipocampo e corpo estriado do cérebro para apoiar a sobrevivência neuronal e melhorar funções motoras.
Propriedades farmacodinâmicas
Sistema imune
Efeitos sobre os números de células imunes no sangue.
Dentro de 4-6 horas após a primeira dose de 0,5 mg de fingolimode, a contagem de linfócitos reduz para aproximadamente 75% do valor basal. Com a continuação da dosagem diária, a contagem de linfócitos continua a reduzir ao longo de um período de duas semanas, alcançando uma contagem nadir de aproximadamente 500 células/?L ou aproximadamente 30% do valor basal.
Dezoito por cento dos pacientes alcançaram um nadir ? 200 células/?L em pelo menos uma ocasião.
Baixas contagens de linfócitos são mantidas com a dosagem crônica diária. A maioria dos linfócitos T e B regularmente circula através dos órgãos linfoides e essas são as células principalmente afetadas pelo fingolimode.
Aproximadamente 15-20% dos linfócitos T possuem um fenótipo de memória efetora, células que são importantes para a vigilância periférica imune.
Uma vez que esse subgrupo de linfócitos geralmente não circula para os órgãos linfoides, ele não é afetado pelo fingolimode.
Aumentos na contagem de linfócitos periféricos são evidentes dentro de dias após a interrupção do tratamento com fingolimode e, geralmente, contagens normais são atingidas dentro de um a dois meses.
A dosagem crônica de fingolimode leva a uma leve redução na contagem de neutrófilos para aproximadamente 80% do valor basal. Monócitos não são afetados pelo fingolimode.
Frequência e ritmo cardíaco
O fingolimode causa uma redução temporária na frequência cardíaca e na condução atrioventricular no início do tratamento.
O declínio máximo da frequência cardíaca é observado nas primeiras 4-5 horas pós-dose, com 70% do efeito cronotrópico negativo atingido no primeiro dia.
A frequência cardíaca retorna progressivamente aos valores basais dentro de um mês do tratamento crônico.
Respostas autonômicas do coração, incluindo a variação diurna da frequência cardíaca e a resposta ao exercício, não são afetadas pelo tratamento com fingolimode.
Com o início do tratamento com fingolimode, ocorre um aumento nas contrações atriais prematuras, mas não há qualquer taxa aumentada de fibrilação/flutter atrial ou arritmias ventriculares ou ectopia.
O tratamento com fingolimode não está associado com a redução no débito cardíaco.
A redução na frequência cardíaca induzida pelo fingolimode pode ser revertida pela atropina, isoprenalina ou salmeterol.
Potencial para prolongar o intervalo QT
Em um estudo minucioso sobre o intervalo QT das doses de 1,25 ou 2,5 mg de fingolimode em estado de equilíbrio, quando um efeito cronotrópico negativo de fingolimode ainda estava presente, o tratamento com fingolimode resultou em um prolongamento do QTcI, com o limite superior do IC de 90% ? 13,0 msec. Não há relação da dose-resposta ou da exposição-resposta de fingolimode com prolongamento de QTcI.
Não há sinais consistentes da incidência aumentada de valores discrepantes no QTcI, sejam eles absolutos ou alterados em relação ao valor basal, associados com o tratamento com fingolimode.
Nos estudos de esclerose múltipla, não houve qualquer prolongamento clinicamente relevante do intervalo QT.
Função pulmonar
O tratamento com fingolimode com dose única ou doses múltiplas de 0,5 e 1,25 mg por duas semanas não está associado com um aumento detectável na resistência das vias aéreas, medida pelo VEF1 e fluxo expiratório forçado durante a expiração de 25 a 75% da capacidade vital forçada (CVF25-75).
No entanto, doses únicas de fingolimode ? 5 mg (10 vezes a dose recomendada) estão associadas a um aumento dose-dependente da resistência das vias aéreas.
O tratamento com fingolimode com doses múltiplas de 0,5, 1,25, ou 5 mg não está associado a oxigenação prejudicada ou dessaturação de oxigênio com exercícios ou aumento da responsividade das vias aéreas à metacolina.
Pacientes submetidos ao tratamento com fingolimode têm resposta broncodilatadora normal aos ?-agonistas inalados.
Propriedades Farmacocinéticas
Absorção
A absorção do fingolimode é lenta (tmáx de 12-16 horas) e extensiva (? 85%, baseados na quantidade de radioatividade excretada na urina e na quantidade de metabólitos extrapolados nas fezes para o infinito).
A biodisponibilidade oral absoluta aparente é alta (93%).
A ingestão de alimentos não altera a Cmáx ou a exposição (AUC) de fingolimode ou fingolimode-fosfato. Portanto, Cloridrato de Fingolimode (substância ativa) pode ser tomado independentemente das refeições.
Concentrações sanguíneas em estado de equilíbrio são atingidas dentro de 1 a 2 meses da administração de uma vez ao dia e os níveis em estado de equilíbrio são aproximadamente 10 vezes maiores do que com a dose inicial.
Distribuição
O fingolimode se distribui altamente nos glóbulos vermelhos, com a fração de 86% nas células sanguíneas.
O fingolimode-fosfato apresenta uma menor captação em células sanguíneas de < 17%. O fingolimode e o fingolimodefosfato são altamente ligados a proteínas (> 99,7%).
A ligação proteica de fingolimode e fingolimode-fosfato não é alterada por danos renais ou hepáticos.
O fingolimode é extensivamente distribuído aos tecidos do corpo com um volume de distribuição de cerca de 1200 ± 260 L.
Um estudo realizado em quatro indivíduos saudáveis que receberam uma única dose intravenosa de fingolimode marcado com radio iodo demonstraram que o fingolimode penetra no cérebro.
Num estudo com 13 pacientes do sexo masculino com esclerose múltipla que receberam Cloridrato de Fingolimode (substância ativa) 0,5 mg/dia, no estado de equilíbrio, a quantidade de fingolimode (e fingolimode-fosfato) na ejaculação seminal era mais do que 10000 vezes menor do que a dose administrada (0,5 mg).
Metabolismo
A biotransformação de fingolimode em humanos ocorre por três vias principais; por fosforilação estereoseletiva reversível para o (S)-enantiômero farmacologicamente ativo de fingolimode-fosfato, por biotransformação oxidativa catalisada principalmente pela CYP4F2 e possivelmente outras isoenzimas CYP4F e subsequente degradação semelhante à do ácido graxo para metabólitos inativos, e pela formação de análogos de ceramida não polares farmacologicamente inativos de fingolimode.
Após a administração oral única de [14C]-fingolimode, os principais componentes no sangue relacionados ao fingolimode, conforme julgados pela sua contribuição à AUC de até 816 horas pós-dose do total de componentes radiomarcados, são o próprio fingolimode (23,3%), fingolimode-fosfato (10,3%), e metabólitos inativos (metabólito do ácido carboxílico M3 (8,3%), metabólito da ceramida M29 (8,9%) e metabólito da ceramida M30 (7,3%)).
Eliminação
O clearance sanguíneo de fingolimode é de 6,3 ±2,3 L/h, e a meia-vida (t1/2) terminal aparente média é de 6-9 dias.
Os níveis sanguíneos de fingolimode-fosfato reduzem em paralelo com fingolimode na fase terminal, produzindo meiasvidas semelhantes para ambos.
Após administração oral, cerca de 81% da dose é lentamente excretada na urina na forma de metabólitos inativos.
O fingolimode e o fingolimode-fosfato não são excretados intactos na urina, mas são os principais componentes nas fezes, com quantidades representando menos que 2,5% da dose, cada.
Após 34 dias, a recuperação da dose administrada é de 89%.
Linearidade
As concentrações de fingolimode e de fingolimode-fosfato aumentam de uma maneira aparentemente proporcional à dose após múltiplas doses de uma vez ao dia de 0,5 mg ou 1,25 mg de fingolimode.
Populações Especiais
Disfunção Renal
O comprometimento renal grave aumenta a Cmáx e a AUC de fingolimode em 32% e 43%, respectivamente, e a Cmáx e a AUC de fingolimode-fosfato em 25% e 14%, respectivamente.
A meia-vida de eliminação aparente não é alterada para ambos os analitos. Nenhum ajuste de dose de Cloridrato de Fingolimode (substância ativa) é necessário em pacientes com comprometimento renal.
Disfunção Hepática
A farmacocinética do fingolimode em dose única (1 ou 5 mg), quando avaliada em indivíduos com comprometimentos hepáticos leve, moderado e grave (Child-Pugh classe A, B e C), não demonstrou qualquer alteração na Cmáx de fingolimode, mas um aumento na AUC em 12%, 44% e 103%, respectivamente.
A meia-vida de eliminação aparente não é alterada pelo comprometimento hepático leve, mas é prolongada em 49-50% pelo comprometimento hepático moderado e grave.
Em pacientes com insuficiência hepática grave (Child-Pugh classe C), o Cmáx do fingolimode-fosfato foi reduzido em 22% e a AUC aumentada em 38%.
A farmacocinética do fingolimode-fosfato não foi avaliada em pacientes com insuficiência hepática leve a moderada.
Embora o comprometimento hepático tenha provocado alterações na disposição de fingolimode e fingolimode-fosfato, a magnitude dessas alterações sugere que a dose de fingolimode não precisa ser ajustada em pacientes com comprometimento hepático leve ou moderado (Child-Pugh classe A e B).
O fingolimode deve ser usado com cautela em pacientes com comprometimento hepático grave (Child-Pugh classe C).
Pediatria
A segurança e eficácia de Cloridrato de Fingolimode (substância ativa) em pacientes pediátricos abaixo de 18 anos não foram estudadas.
Cloridrato de Fingolimode (substância ativa) não é indicado para o uso em pacientes pediátricos.
Geriatria
O mecanismo de eliminação e os resultados da população farmacocinética sugerem que o ajuste de dose não seria necessário em pacientes idosos. Entretanto, a experiência clínica em pacientes com mais de 65 anos de idade é limitada.
Etnia
Os efeitos de origem étnica na farmacocinética de fingolimode e fingolimode-fosfato não são de relevância clínica.
Sexo
O sexo não exerce influência sobre a farmacocinética de fingolimode e fingolimode-fosfato.
Dados de segurança pré-clínicos
O perfil de segurança pré-clínico de fingolimode foi avaliado em camundongos, ratos, cães e macacos.
Os principais órgãos-alvo foram o sistema linfoide (linfopenia e atrofia linfoide), pulmões (aumento de peso, hipertrofia do músculo liso na junção bronquioalveolar), e coração (efeito cronotrópico negativo, aumento na pressão arterial, alterações perivasculares e degeneração do miocárdio) em diversas espécies; vasos sanguíneos (vasculopatia) apenas em ratos; e pituitária, pré-estômago, fígado, adrenais, trato gastrintestinal e sistema nervoso apenas em altas doses (frequentemente associados com sinais de toxicidade geral) em diversas espécies.
Nenhuma evidência de carcinogenicidade foi observada em um bioensaio de 2 anos em ratos em doses orais de fingolimode até a dose máxima tolerada de 2,5 mg/kg, representado uma margem de aproximadamente 50 vezes com base na exposição sistêmica humana (AUC) na dose de 0,5 mg.
Entretanto, em um estudo de 2 anos em camundongos, uma incidência elevada de linfoma maligno foi observada em doses de 0,25 mg/kg e superiores, representando uma margem de aproximadamente 6 vezes com base na exposição sistêmica humana (AUC) em uma dose diária de 0,5 mg.
O fingolimode não foi mutagênico em um teste Ames e em uma linhagem celular de linfoma L5178Y de camundongo in vitro.
Nenhum efeito clatogênico foi observado in vitro em células pulmonares V79 de hamster chinês. O fingolimode induziu aberrações cromossômicas numéricas (poliploide) em células V79 em concentrações de 3,7 mcg/mL e superiores. O fingolimode não foi clastogênico nos teste de micronúcleo in vivo em camundongos e ratos.
O fingolimode não teve qualquer efeito na contagem ou motilidade de esperma, nem na fertilidade em ratos machos e fêmeas até a dose mais alta testada (10 mg/kg), representando uma margem de aproximadamente 150 vezes com base na exposição sistêmica humana (AUC) em uma dose diária de 0,5 mg.
Em um estudo de toxicidade em ratos jovens, nenhum órgão-alvo adicional de toxicidade foi observado em comparação com os ratos adultos. Estímulos repetidos com hemocianina do molusco lapa californiana (KLH) demonstraram uma resposta moderadamente reduzida durante o período de tratamento, mas reações imunes totalmente em funcionamento no final de um período de recuperação de 8 semanas.
O fingolimode foi excretado no leite de animais tratados durante a lactação. O fingolimode e seus metabólitos cruzaram a barreira placentária em coelhas prenhas.
Cuidados de Armazenamento
Gilenya deve ser mantido em temperatura ambiente (entre 15 e 30°C) e protegido da umidade.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas
Cápsulas com corpo branco opaco e tampa amarela clara opaca, contendo pó branco a quase branco.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças
Mensagens de Alerta
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Dizeres Legais
MS – 1.0068.1076
Farm. Resp.: Flavia Regina Pegorer – CRF-SP 18.150
Importado por:
Novartis Biociências S.A.
Av. Prof. Vicente Rao, 90. São Paulo – SP.
CNPJ:56.994.502/0001-30
Indústria Brasileira
Fabricado por:
Novartis Pharma Stein AG, Stein, Suíça