Comparamos o preço de Myfortic 360Mg C 120 Comprimidos, veja o menor preço

R$ 2.858,70
RReferência
12
ofertasMelhores preços a partir de R$ 2.858,70 até R$ 3.500,00
Menor preço
vendido por Farma Visa
economize
18.32%
R$ 2.858,70
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
Menor preço
vendido por Life Medicamentos
economize
18.32%
R$ 2.858,70
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Imune Farma Medicamentos Especiais
economize
18.32%
R$ 2.858,80
vendido por Farma Ame
economize
18.32%
R$ 2.858,85
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Farma Silva
economize
15.83%
R$ 2.945,80
Frete Grátis para SP e MG em Compras Acima de R$ 149,90.
vendido por Drogaria Dinâmica
economize
13.40%
R$ 3.030,98
Dúvidas quanto ao Preço ou Frete? Clique e vá direto ao site da Drogaria.
vendido por Facilita Medicamentos
economize
13.07%
R$ 3.042,54
vendido por Drogaria Araújo
economize
7.31%
R$ 3.243,99
vendido por Fast Medicamentos
economize
6.09%
R$ 3.286,70
vendido por Onco Express Medicamentos Especiais e Oncológicos
economize
0.29%
R$ 3.490,00
Farmácia de medicamentos oncológicos. Parcelamos em até 6 x sem juros.
vendido por OncoExpresso Medicamentos
economize
0.29%
R$ 3.490,00
vendido por Maranata Medicamentos
R$ 3.500,00
Para que serve
Myfortic é usado para prevenir a rejeição ao transplante renal. É usado juntamente com outros medicamentos contendo ciclosporina e corticoides.
Se você tem alguma dúvida sobre como Myfortic funciona ou por que este medicamento foi prescrito para você, pergunte ao seu médico.
Como o Myfortic funciona?
Myfortic comprimidos revestidos gastrorresistentes pertence à classe de medicamentos conhecida como imunossupressores. Os imunossupressores reduzem a resposta do corpo a agentes reconhecidos como “estranhos” – assim como órgãos transplantados.
Contraindicação
- Se você for alérgico (hipersensível) ao ácido micofenólico, micofenolato de sódio, micofenolato de mofetila ou a qualquer um dos excipientes listados no item “Composição”;
- Se você suspeitar que seja alérgico, solicite orientação do seu médico.
Se alguma das situações acima se aplicar a você, informe ao seu médico antes de tomar Myfortic.
Se você acha que pode ser alérgico, converse com seu médico.
Como usar
Siga cuidadosamente as instruções do seu médico. Não exceda a dose recomendada.
Quando e como tomar Myfortic
Engula os comprimidos inteiros com um copo de água.
Não quebre ou amasse os comprimidos.
Não tome nenhum comprimido que esteja quebrado.
Quanto tomar de Myfortic
A dose diária recomendada é 1.440 mg (8 comprimidos de Myfortic 180 mg ou 4 comprimidos de Myfortic 360 mg), tomada em 2 doses separadas de 720 mg cada, de estômago vazio, 1 hora antes ou duas horas após a ingestão de alimento.
Isto significa tomar 4 comprimidos de Myfortic 180 mg ou 2 comprimidos de Myfortic 360 mg pela manhã e 4 comprimidos de Myfortic 180 mg ou 2 comprimidos de Myfortic 360 mg à noite.
A primeira dose de 720 mg será dada dentro de 48 horas após o transplante.
Seu médico vai dizer exatamente quantos comprimidos de Myfortic tomar.
Por quanto tempo tomar Myfortic
O tratamento continuará enquanto você precisar de imunossupressão para prevenir a rejeição do seu rim transplantado.
Se você parar de tomar Myfortic
A interrupção do tratamento com Myfortic pode aumentar o risco de rejeição do rim transplantado. Não pare de tomar o seu medicamento, a menos que seu médico lhe diga.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
O que fazer quando eu me esquecer de usar o Myfortic?
Se você se esquecer de tomar Myfortic, tome-o assim que se lembrar, então, continue a tomá-lo nos horários habituais.
Pergunte ao seu médico.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Myfortic somente será prescrito para você por um médico com experiência em transplantes.
Siga cuidadosamente as instruções do seu médico. Elas podem diferir da informação geral contida nesta bula.
Tome cuidado especial com Myfortic:
- Durante exposição à luz solar. Myfortic reduz o mecanismo de defesa do seu corpo, o que pode aumentar o risco de câncer de pele. Desta forma, você deve limitar sua exposição à luz solar e à luz UV (ultravioleta), utilizando roupa protetora apropriada e aplicando frequentemente protetor solar com um alto fator de proteção;
- Se você já teve hepatite B ou C, Myfortic pode aumentar o risco de estas doenças reaparecerem. Seu médico poderá realizar análises sanguíneas e verificar se há sintomas destas doenças. Se você apresentar quaisquer sintomas (pele e olhos amarelados, náuseas, perda de apetite, urina escura), você deve informar ao seu médico imediatamente;
- Se você apresentar qualquer sintoma de infecção (por exemplo, febre, dor de garganta), lesão e/ou sangramentos inesperados. Neste caso você deve informar imediatamente ao seu médico;
- Se precisar ser vacinado consulte o seu médico antes;
- Se você tem ou teve um distúrbio grave do trato digestivo, como por exemplo úlcera estomacal;
- Se você tem uma deficiência hereditária rara da enzima hipoxantina-guanina fosforibosil-transferase (HGPRT), como a síndrome de Lesch-Nyhan (também conhecida como síndrome de Kelley-Seegmiller);
- O uso de Myfortic em mulheres grávidas pode aumentar o risco de defeitos no bebê e perda da gravidez, incluindo aborto espontâneo. Se você for mulher e estiver em idade fértil o tratamento com Myfortic não deve ser iniciado até que seja realizado um teste com resultado negativo para gravidez e você deve utilizar método contraceptivo durante o tratamento e por pelo menos 6 semanas após o término do tratamento;
- Se você estiver grávida, desconfia que esteja grávida ou pretende engravidar, consulte seu médico;
- Se você estiver amamentando.
Se alguma das situações acima se aplicar a você, informe ao seu médico antes de tomar Myfortic.
Reações Adversas
Como todo medicamento, Myfortic pode causar reações adversas, embora nem todas as pessoas as apresentem.
Algumas das mais frequentes são
- Constipação;
- Diarreia;
- Náuseas;
- Infecções;
- Diminuição de células brancas em seu sangue.
O seu médico fará exames de sangue regulares para monitorar qualquer alteração no número de suas células sanguíneas ou alterações nos níveis de substâncias presentes em seu sangue, como açúcar, colesterol e lipídeos.
Algumas reações adversas podem ser graves
Caso apresente qualquer um desses sinais ou sintomas, informe imediatamente ao seu médico.
- Se você apresentar sintomas de infecção, incluindo febre, calafrio, suor, sensação de cansaço, sonolência ou falta de energia. Se você está tomando Myfortic você pode estar mais susceptível a infecções do que o usual. Estas podem ocorrer em vários sistemas do seu corpo, mas são mais comuns no trato urinário, sistema respiratório e pele;
- Se você apresentar alterações visuais, perda de coordenação, perda de memória, dificuldade na fala ou compreensão e fraqueza muscular. Esses podem ser sinais e sintomas de uma infecção do cérebro denominada leucoencefalopatia multifocal progressiva;
- Se você apresentar glândulas aumentadas, crescimento novo ou aumentado de pele ou uma alteração de uma pinta existente. Como pode ocorrer em pacientes sob tratamento imunossupressor, um número muito pequeno de pacientes tratados com Myfortic desenvolveu câncer de pele ou nódulos linfáticos;
- Se você apresentar cansaço não usual, dor de cabeça, falta de ar com exercício ou em repouso, tontura, dor no peito, palidez. Esses são sintomas de anemia (redução de células vermelhas do sangue).
Outras reações adversas podem incluir
Algumas reações adversas são muito comuns (ocorrem em mais de 10% dos pacientes que utilizam este medicamento)
- Baixo nível de células brancas no sangue;
- Nível de cálcio reduzido no sangue, algumas vezes com cólicas (hipocalcemia);
- Fraqueza muscular, espasmos musculares, ritmo cardíaco anormal (possíveis sintomas de nível baixo de potássio no sangue) (hipocalemia);
- Resultados anormais nos exames de sangue (nível alto de ácido úrico no sangue) (hiperuricemia);
- Dor de cabeça, tontura (possíveis sintomas de pressão sanguínea alta) (hipertensão);
- Tontura, delírio (possíveis sintomas de pressão sanguínea baixa) (hipotensão);
- Diarreia.
Algumas reações adversas comuns (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento)
- Hemorragia ou hematomas com mais facilidade do que o normal (sinais de baixo nível de plaquetas no sangue);
- Espasmos musculares, ritmo cardíaco anormal (possíveis sintomas de nível alto de potássio no sangue) (hipercalemia);
- Resultados anormais nos exames de sangue (nível baixo de magnésio no sangue) (hipomagnesemia);
- Excessiva aflição emocional, preocupação (sintomas de ansiedade);
- Tontura;
- Dor de cabeça;
- Tosse;
- Dor de cabeça, tontura, possivelmente com náuseas (possíveis sintomas graves de pressão sanguínea alta (piora da hipertensão);
- Falta de ar, respiração com dificuldade (possíveis sintomas de dispneia ou dispneia de esforço);
- Dor (por exemplo, no abdômen, estômago);
- Constipação;
- Indigestão;
- Flatulência
- Fezes amolecidas;
- Náuseas;
- Vômitos;
- Cansaço;
- Febre;
- Resultados anormais no teste de função hepática ou renal;
- Dor nas articulações (artralgia);
- Fraqueza (astenia);
- Dor muscular (mialgia);
- Inchaço nas mãos, tornozelos ou pés (possíveis sintomas de edema periférico).
Algumas reações adversas incomuns (ocorre em menos de 1% dos pacientes que utilizam este medicamento)
- Cisto contendo líquido linfático;
- Dificuldade em dormir, tremores;
- Congestão pulmonar;
- Falta de ar;
- Arrotos;
- Mau hálito;
- Obstrução intestinal;
- Inflamação do esôfago;
- Fezes escuras ou com sangue;
- Boca seca, lesões no lábio;
- Bloqueio das glândulas salivares, azia, inflamação das gengivas, inflamação do revestimento da cavidade abdominal;
- Sintomas gripais, calafrios;
- Inchaço dos tornozelos e pés;
- Perda de apetite;
- Dor nas costas, dor muscular;
- Queda de cabelo;
- Equimose da pele;
- Acne;
- Batimento cardíaco acelerado;
- Cansaço dos olhos com coceira, vermelhidão e inchaço, visão turva;
- Ideias delirantes;
- Distúrbios renais, estreitamento anormal do tubo pelo qual a urina passa para fora do corpo, sangue na urina;
- Tosse, dificuldade em respirar, dor ao respirar (possíveis sintomas de doença pulmonar intersticial incluindo fibrose pulmonar fatal).
Outras reações adversas tem frequência desconhecida (a frequência não pode ser estimada a partir dos dados disponíveis)
- Rash;
- Febre, dor de garganta, infecções frequentes (possíveis sintomas da falta de células brancas no sangue) (agranulocitose).
Outras reações adversas reportadas com medicamentos semelhantes ao Myfortic
Reações adversas adicionais foram relatadas com a classe de medicamentos a qual Myfortic pertence
- Inflamação do cólon ou do esôfago;
- Dor abdominal, vômitos, perda de apetite, náuseas (inflamação do pâncreas);
- Perfuração intestinal;
- Sangramento do estômago ou intestino;
- Dor de estômago com ou sem fezes escuras ou com sangue;
- Obstrução intestinal;
- Infecções graves;
- Redução do número de células brancas específicas ou de todas as células do sangue.
Se alguma destas reações adversas afetar você, informe ao seu médico.
Caso apresente outras reações adversas não mencionadas nesta bula, informe ao seu médico ou farmacêutico. No entanto, não pare de tomar seus medicamentos a menos que tenha discutido isso com o seu médico.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento.
População Especial
Mulheres em idade fértil
Seu médico deve orientá-la quanto ao uso de contraceptivos antes de iniciar o tratamento com Myfortic.
Você deve utilizar contraceptivo antes e durante o tratamento com Myfortic e por 6 semanas após você ter parado de tomar Myfortic.
Fale com seu médico imediatamente se você engravidar durante o tratamento com Myfortic.
Gravidez
O uso de Myfortic na gravidez pode aumentar o risco de defeitos de nascença e de perda da gravidez. Se você estiver grávida ou achar que pode estar, ou planeja engravidar, informe ao seu médico. O seu médico irá discutir com você os riscos potenciais de tomar Myfortic durante a gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Amamentação
Informe ao seu médico se estiver amamentando.
Não amamente durante o tratamento com Myfortic e por até 6 semanas após você ter parado de tomar Myfortic.
Homens
Se você é homem sexualmente ativo, você deve utilizar preservativos (camisinha) durante o tratamento com Myfortic e por 13 semanas após o término do tratamento.
Sua parceira também deve utilizar um método contraceptivo efetivo durante o seu tratamento com Myfortic e por 13 semanas após o término do seu tratamento.
Informe seu médico imediatamente se sua parceira engravidar enquanto você estiver tomando Myfortic.
Idosos
Myfortic pode ser usado por idosos. Não é necessário ajuste de dose.
Crianças e adolescentes
A experiência com Myfortic em crianças é muito limitada.
Efeitos na habilidade de dirigir veículos e/ou operar máquinas
Não é provável que Myfortic afete a habilidade de dirigir veículos e/ou operar máquinas.
Composição
Myfortic 180 mg
Cada comprimido contém 180 mg de ácido micofenólico equivalente a 192,4 mg de micofenolato de sódio.
Excipientes: amido, povidona, crospovidona, lactose, dióxido de silício, estearato de magnésio, ftalato de hipromelose, dióxido de titânio, óxido férrico amarelo e indigotina.
Myfortic 360 mg
Cada comprimido contém 360 mg de ácido micofenólico equivalente a 384,8 mg de micofenolato de sódio.
Excipientes: amido, povidona, crospovidona, lactose, dióxido de silício, estearato de magnésio, ftalato de hipromelose, dióxido de titânio, óxido férrico amarelo e óxido férrico vermelho.
Superdosagem
Se você tomar acidentalmente muitos comprimidos, informe ao seu médico imediatamente. Você pode precisar de cuidados médicos.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Aciclovir:
Concentrações plasmáticas maiores de aciclovir e MPAG foram observadas quando micofenolato de mofetila foi administrado com aciclovir em comparação com a administração de cada droga isoladamente. Devido ao aumento da concentração plasmática de MPAG na presença de disfunção renal, como ocorre com o aciclovir, pode ocorrer competição entre o micofenolato de mofetila e o aciclovir ou suas pró-drogas como o valganciclovir pela secreção tubular e isso pode aumentar as concentrações de ambas as drogas.
Antiácidos e inibidores da bomba de prótons (IBPs):
Exposição diminuída de ácido micofenólico (MPA) foi observada quando antiácidos, como hidróxidos de alumínio e magnésio, e IBPs, incluindo omeprazol, lansoprazol e pantoprazol, foram administrados com micofenolato de mofetila. Não foram observadas diferenças significativas ao comparar as taxas de rejeição de transplante ou as taxas de perda do enxerto entre pacientes de micofenolato de mofetila que utilizaram IBPs versus pacientes de micofenolato de mofetila que não utilizaram IBPs. Esses dados suportam a extrapolação dessa observação para todos os antiácidos, pois a redução à exposição é consideravelmente menor quando micofenolato de mofetila foi coadministrado com hidróxidos de alumínio e magnésio em relação à coadministração com IBPs.
Colestiramina:
Após administração de 1,5 g de micofenolato de mofetila em indivíduos saudáveis pré-tratados com colestiramina 4 g, três vezes ao dia durante quatro dias, houve redução de 40% na AUC do MPA. Deve-se ter cautela durante a administração concomitante de drogas que interfiram na circulação entero-hepática.
Ciclosporina A:
A farmacocinética da ciclosporina A (CsA) não é afetada por micofenolato de mofetila. Entretanto, CsA interfere na circulação entero-hepática do MPA, resultando em reduções de 30% a 50% na exposição do MPA em pacientes transplantados renais tratados com micofenolato de mofetila e CsA, se comparado com pacientes que receberam sirolimo ou belatacept e doses semelhantes de micofenolato de mofetila. Inversamente, mudanças na exposição de MPA devem ser esperadas quando há substituição do uso de CsA para um dos imunossupressores que não interferem na circulação entero- hepática do MPA.
Telmisartana:
A administração concomitante de telmisartana e micofenolato de mofetila resultou em uma diminuição de aproximadamente 30% nas concentrações de ácido micofenólico (MPA). Telmisartan altera a eliminação do MPA por aumentar a expressão de PPAR gama (receptor gama ativador da proliferação de peroxissoma), que por sua vez resulta no aumento da expressão e da atividade de UGT1A9. Ao comparar as taxas de rejeição do transplante, as taxas de perda do enxerto ou perfis de eventos adversos entre os pacientes de micofenolato de mofetila com e sem uso concomitante de telmisartana, não foram observadas consequências clínicas da interação medicamentosa farmacocinética.
Ganciclovir:
baseado nos resultados de um estudo com administração de dose única, nas doses recomendadas de o micofenolato de mofetila oral e ganciclovir intravenoso e nos efeitos conhecidos da deterioração renal sobre a farmacocinética do MMF e do ganciclovir, prevê-se que a co-administração desses agentes (que competem pelos mecanismos de secreção tubular renal) resultará em aumento na concentração do MPAG e do ganciclovir. Nenhuma alteração substancial na farmacocinética do MPA é prevista, não sendo necessário o ajuste da dose do MMF.
Pacientes com deterioração renal nos quais o MMF e o ganciclovir ou suas pró-drogas, como o valganciclovir, são co-administrados, devem ser monitorados cuidadosamente.
Contraceptivos orais:
Um estudo de coadministração de micofenolato de mofetila (1 g duas vezes ao dia) e contraceptivo oral combinado contendo etinilestradiol (0,02 - 0,04 mg) e levonorgestrel (0,05 - 0,20 mg), desogestrel (0,15 mg) ou gestodene (0,05 - 0,10 mg) envolvendo 18 mulheres com psoríase e conduzido por mais de três ciclos menstruais não mostrou influência clínica relevante de micofenolato de mofetila nos níveis séricos da progesterona, do LH e do FSH, não indicando, portanto, influência de micofenolato de mofetila no efeito supressor da ovulação dos contraceptivos orais. A farmacocinética dos contraceptivos orais não foi afetada em um nível clinicamente relevante pela co-administração de micofenolato de mofetila.
Rifampicina:
Após correção da dose, uma diminuição em 70% da exposição de MPA (AUC0-12h) foi observada com administração concomitante de rifampicina em um único paciente transplantado de coração e pulmão. Portanto, recomenda-se controlar os níveis de exposição de MPA e ajustar as doses de micofenolato de mofetila para manter a eficácia clínica quando as drogas são administradas concomitantemente.
Tacrolimo:
Não foi observado efeito na AUC ou Cmáx do MPA em pacientes transplantados hepáticos, ao administrar tacrolimo concomitantemente com micofenolato de mofetila. Observou-se resultado similar em um estudo recente, em pacientes transplantados renais.
Em pacientes transplantados renais, mostrou-se que a concentração do tacrolimo parece não ser alterada pelo micofenolato de mofetila.
Entretanto, em pacientes transplantados hepáticos estáveis, observou-se aumento de aproximadamente 20% na AUC do tacrolimo, quando foram administradas doses múltiplas de micofenolato de mofetila (1,5 g duas vezes ao dia) para pacientes recebendo tacrolimo.
Antibióticos eliminadores de bactérias intestinais produtoras de -glucuronidase (por exemplo, aminoglicosídeos, cefalosporinas, fluoroquinolona, e antibióticos penicilínicos) podem interferir na recirculação entero-hepática, o que acarreta na redução da exposição sistêmica de MPA.
Informações acerca dos seguintes antibióticos estão disponíveis:
Ciprofloxacina ou amoxicilina associada ao ácido clavulânico:
Reduções de 54% nas concentrações pré-tomada (vale) de MPA foram relatadas em pacientes transplantados renais nos dias imediatamente após o início da ciprofloxacina oral ou amoxicilina associada ao ácido clavulânico. Os efeitos tendem a diminuir com o uso continuado do antibiótico e a cessar após a descontinuação. A alteração no nível de pré-dose pode não representar exatamente as alterações na exposição global ao MPA, portanto, a relevância clínica dessas observações é incerta.
Norfloxacino e metronidazol:
Norfloxacino em combinação com metronidazol diminuiu a AUC0-48 do MPA em 30%, após dose única de micofenolato de mofetila. Esse efeito sobre a exposição sistêmica de MPA não ocorreu com qualquer um destes antibióticos quando foram administradas separadamente.
Trimetoprima - sulfametoxazol:
Não foi observado efeito sobre a exposição sistêmica do MPA (AUC, Cmax) com a combinação trimetoprima / sulfametoxazol.
Outras interações:
Coadministração de probenecida com micofenolato de mofetila em macacos aumenta, em três vezes, a AUC plasmática do MPAG.
Portanto, outras drogas que sofrem secreção tubular renal podem competir com o MPAG e aumentar a concentração plasmática de ambas.
A administração concomitante de sevelamer e micofenolato de mofetila, em pacientes adultos, diminuiu a Cmáx e a AUC0-12 do MPA em aproximadamente 30% e 25%, respectivamente. Esses dados sugerem que o sevelamer e outros ligantes de fosfato livres de cálcio devem ser administrados, preferencialmente, duas horas após a tomada de micofenolato de mofetila, para minimizar o impacto na absorção do MPA.
Vacinas de vírus vivos:
Vacinas de vírus vivos não devem ser administradas a pacientes com alteração da resposta imune. A resposta de anticorpos a outras vacinas pode estar diminuída.
Interação Alimentícia
A alimentação não teve nenhum efeito sobre a extensão da absorção (AUC do MPA) de micofenolato de mofetila quando administrado na dose de 1,5 g duas vezes ao dia em pacientes submetidos a transplante renal. Porém, a Cmáx do MPA diminuiu 40% na presença de alimento.
Ação da Substância
Resultados da eficácia
Eficácia
O micofenolato de mofetila foi administrado, em estudos clínicos, para a prevenção de episódios de rejeição em transplante renal, cardíaco e hepático, em associação com os seguintes agentes: imunoglobulina antitimocítica, OKT3, ciclosporina A e corticosteroides. O micofenolato de mofetila foi também utilizado, em associação com a ciclosporina A e corticosteroide, para o tratamento de episódios de rejeição refratária em transplante renal. Antes do tratamento com micofenolato de mofetila, o paciente poderia também ter recebido imunoglobulina antilinfocítica, imunoglobulina antitimocítica e OKT3. O micofenolato de mofetila, além disso, foi utilizado em estudos clínicos associado ao daclizumabe e tacrolimo.
Prevenção da rejeição de órgãos
Adultos:
A segurança e a eficácia de micofenolato de mofetila, em associação com corticosteroides e ciclosporina A, para a prevenção da rejeição do enxerto, foram avaliadas em três estudos multicêntricos, randomizados e duplo-cegos em receptores de transplante renal, em um estudo randomizado e duplo-cego em receptores de transplante cardíaco e em um estudo multicêntrico, randomizado e duplo-cego em receptores de transplante hepático.
Transplante renal
Adultos:
Os três estudos compararam duas doses de micofenolato de mofetila oral (1 g, duas vezes ao dia, e 1,5 g, duas vezes ao dia) com a azatioprina (dois estudos) ou placebo (um estudo) quando administrados em associação com ciclosporina A e corticosteroide, para prevenir episódios de rejeição aguda.
O desfecho principal de eficácia foi a proporção de pacientes em cada grupo de tratamento que apresentaram falha de tratamento nos primeiros seis meses após o transplante (definida como rejeição aguda comprovada por biópsia ou a ocorrência de morte, perda do enxerto ou a retirada prematura do estudo por qualquer razão que não rejeição comprovada por biópsia). O micofenolato de mofetila foi avaliado em três esquemas terapêuticos: (1) indução com imunoglobulina antitimocítica/MMF ou azatioprina/ciclosporina A/corticosteroide, (2) MMF ou azatioprina/ciclosporina A/corticosteroide, e (3) MMF ou placebo/ciclosporina A/corticosteroide.
O micofenolato de mofetila, em associação com corticosteroides e ciclosporina A, reduziu a incidência de falha de tratamento (p < 0,05) nos primeiros seis meses após o transplante. As tabelas, a seguir, resumem os resultados destes estudos. Os pacientes que descontinuaram prematuramente o tratamento foram acompanhados quanto à ocorrência de morte ou de perda do enxerto, sendo que a incidência cumulativa destes dois eventos está descrita separadamente. Pacientes que descontinuaram prematuramente o tratamento não foram acompanhados quanto à ocorrência de rejeição aguda após o término. Um número maior de pacientes no grupo micofenolato de mofetila descontinuou o tratamento (sem rejeição comprovada por biópsia, morte ou perda do enxerto prévia), quando comparado com o grupo controle, com os índices mais altos no grupo de micofenolato de mofetila 3 g/dia. Entretanto, os índices de rejeição aguda podem estar subestimados, particularmente no grupo de micofenolato de mofetila 3 g/dia.
Estudos em transplante renal
Incidência de falha de tratamento (Rejeição comprovada por biópsia ou término precoce por qualquer motivo)
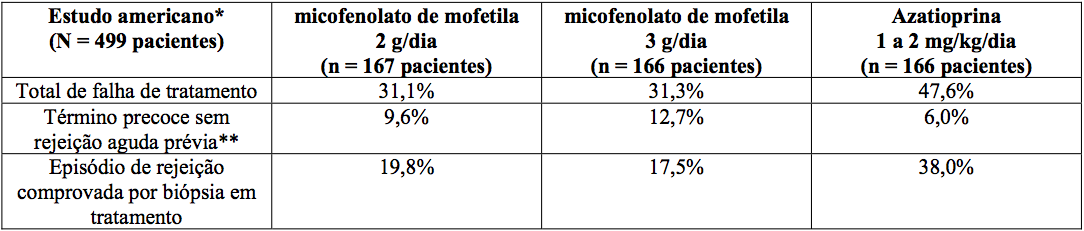
*Indução com imunoglobulina antitimocítica/MMF ou azatioprina/ciclosporina A/corticosteroides.
*MMF ou azatioprina/ciclosporina A/corticosteroides.


*MMF ou placebo/ciclosporina A/corticosteroides.
**Não inclui morte ou perda do enxerto como razão para o término precoce.
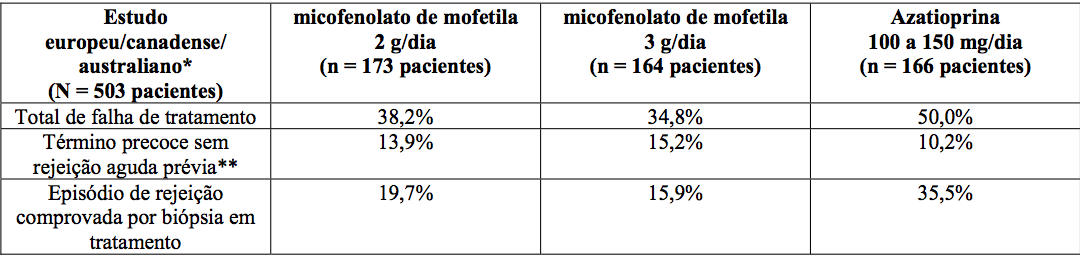
A incidência cumulativa de perda do enxerto e de morte de pacientes aos 12 meses está apresentada a seguir. Nenhuma superioridade de micofenolato de mofetila em relação à perda do enxerto e à morte de paciente foi estabelecida. Numericamente, os pacientes que receberam micofenolato de mofetila 2 g/dia e 3 g/dia apresentaram melhores resultados que os pacientes do grupo controle nos três estudos; pacientes que receberam micofenolato de mofetila 2 g/dia apresentaram melhores resultados que os que receberam 3 g/dia em dois dos três estudos. Em todos os grupos de tratamento, os pacientes que terminaram prematuramente o tratamento tiveram resultados piores em relação à perda do enxerto e à morte de pacientes com um ano.
Estudos em transplante renal
Incidência cumulativa de perda do enxerto e morte de pacientes em 12 meses
Transplante cardíaco
Um estudo multicêntrico, de grupos paralelos, randomizado, comparativo e duplo-cego foi realizado em receptores primários de transplante cardíaco. Foram envolvidos 650 pacientes; 72 não receberam droga do estudo e 578 receberam.
Os pacientes receberam micofenolato de mofetila 1,5 g duas vezes ao dia (n = 289) ou azatioprina 1,5 a 3 mg/kg/dia (n = 289), em associação com ciclosporina A e corticosteroide como terapia imunossupressora de manutenção. Os dois parâmetros primários de eficácia foram: (1) a proporção de pacientes que, após o transplante, apresentaram pelo menos um episódio de rejeição comprovada por biópsia endomiocárdica, com comprometimento hemodinâmico ou foram retransplantados ou morreram nos primeiros seis meses, e (2) a proporção de pacientes que morreram ou foram retransplantados nos primeiros 12 meses após o transplante. Os pacientes que descontinuaram prematuramente o tratamento foram acompanhados quanto à ocorrência de rejeição do enxerto por até seis meses e quanto à ocorrência de morte por um ano.
Rejeição:
Nenhuma diferença foi estabelecida entre micofenolato de mofetila e azatioprina em relação à rejeição comprovada por biópsia com comprometimento hemodinâmico, como apresentado abaixo.
Rejeição em seis meses?
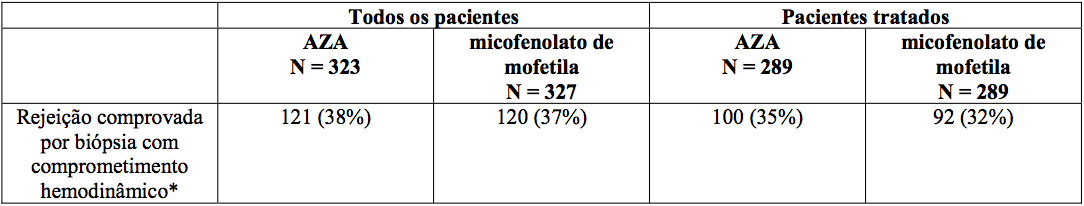
*Comprometimento hemodinâmico ocorreu quando os seguintes critérios foram encontrados: gradiente de pressão capilar pulmonar 20 mm ou um aumento de 25%; índice cardíaco < 2,0 L/min/m2 ou uma diminuição de 25%; fração de ejeção 30%; saturação de oxigênio da artéria pulmonar 60% ou uma diminuição de 25%; presença de ritmo de galope com B3; restrição de fração 20% ou uma diminuição de 25%; necessidade de suporte inotrópico para controle das condições clínicas.
Sobrevida:
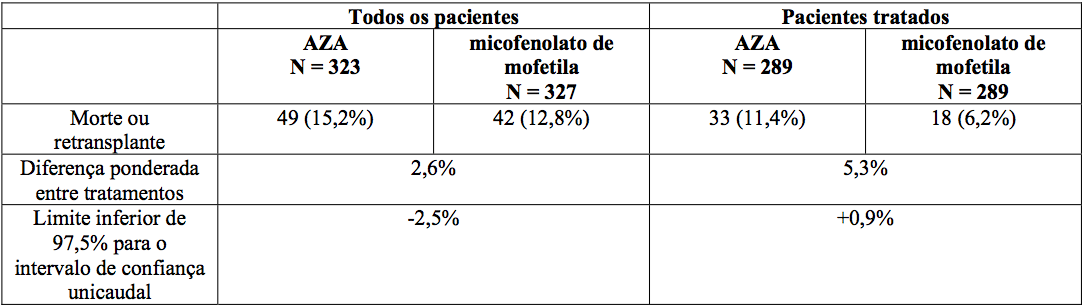
Nos pacientes envolvidos no estudo não houve diferença estatisticamente significativa em relação à morte e retransplante entre os pacientes randomizados do grupo micofenolato de mofetila e os do grupo azatioprina. Nos pacientes que receberam droga do estudo, o limite inferior de 97,5% para o intervalo de confiança da diferença entre morte e retransplante foi de 0,9 no primeiro ano, indicando que micofenolato de mofetila foi superior à azatioprina nesses pacientes, como apresentado abaixo.
Morte ou retransplante no primeiro ano
Transplante hepático
Um estudo multicêntrico, paralelo, randomizado, comparativo e duplo-cego em receptores primários de transplante hepático foi realizado em 16 centros nos EUA, em dois no Canadá, em quatro na Europa e em um na Austrália. O número total de pacientes envolvidos foi de 565, sendo que 564 receberam os medicamentos do estudo. Os pacientes receberam micofenolato de mofetila, 1 g, duas vezes ao dia IV, por 14 dias, seguido de micofenolato de mofetila, 1,5 g, duas vezes ao dia por via oral ou azatioprina 1 - 2 mg/kg/dia IV, seguida por azatioprina 1 - 2 mg/kg/dia por via oral, em associação com ciclosporina A e corticosteroide como terapia imunossupressora. Os dois parâmetros principais de eficácia foram: (1) a proporção de pacientes que apresentaram, nos primeiros seis meses após o transplante, um ou mais episódios de rejeição tratada comprovada por biópsia ou morte/retransplante, e (2) a proporção de pacientes que apresentaram perda do enxerto (morte/retransplante) nos primeiros 12 meses após o transplante. Os pacientes que descontinuaram prematuramente o tratamento foram acompanhados quanto à ocorrência de rejeição do enxerto e quanto à ocorrência de perda do enxerto (morte/retransplante) por um ano.
Resultados:
Em uma análise primária (intenção de tratamento), micofenolato de mofetila, em associação com ciclosporina A e corticosteroide, foi superior à azatioprina na prevenção da rejeição aguda (p = 0,025) e equivalente à azatioprina em relação à sobrevida.
Rejeição em seis meses/morte ou retransplante em um ano
| AZA N = 287 | Micofenolato de mofetila N = 278 | |
| Rejeição tratada comprovada por biópsia em seis meses | 137 (47,7%) | 107 (38,5%) |
| Morte ou retransplante em um ano | 42 (14,6%) | 41 (14,7%) |
Tratamento da rejeição refratária
Um estudo randomizado, aberto e comparativo de MMF 3 g/dia versus corticosteroide intravenoso foi realizado em 150 receptores de transplante renal com rejeição aguda e refratária do enxerto. O parâmetro principal foi a proporção de pacientes que permaneceram vivos e com enxerto funcionante após seis meses da entrada no estudo.
Resultados:
A incidência de perda do enxerto foi inesperadamente pequena no grupo controle e a análise primária, baseada no teste da taxa de probabilidade sequencial, mostrou uma tendência de maior sobrevida do enxerto no grupo MMF (p = 0,081). Uma análise secundária, usando o teste de Cochran-Mantel-Haenzel (não ajustado para o monitoramento sequencial), sugeriu uma redução de 45% na incidência de perda do enxerto ou morte no grupo MMF após seis meses da entrada no estudo (p = 0,062).
Perda do enxerto ou morte em seis meses
| Corticoide IV N = 73 | Micofenolato de mofetila N = 77 | |
| Perda do enxerto ou morte em seis meses | 19 (26,0%) | 11 (14,3 |
Características Farmacológicas
Farmacodinâmica
O micofenolato de mofetila (MMF) é o éster 2-morfolinoetil do ácido micofenólico (MPA). MPA é um inibidor potente, seletivo, não-competitivo e reversível da inosina monofosfato desidrogenase (IMPDH) e, portanto, inibe a via de novo da síntese do nucleotídeo guanosina sem incorporação ao DNA. O mecanismo pelo qual o MPA inibe a atividade enzimática da IMPDH parece estar relacionado à capacidade do MPA em mimetizar estruturalmente tanto o co-fator dinucleotídeo adenina nicotinamida, como uma molécula catalítica de água. Isso impede a oxidação do IMP a xantose-5’-monofosfato, que é um passo fundamental na síntese de novo do nucleotídeo guanosina. O MPA tem efeito citostático maior nos linfócitos que em outras células, pois os linfócitos T e B são extremamente dependentes, para a sua proliferação, da via de novo da síntese das purinas, ao passo que outras células podem utilizar vias alternativas.
Farmacocinética
A farmacocinética do MMF foi estudada em pacientes de transplante renal, cardíaco e hepático. Em geral, o perfil farmacocinético do MPA é semelhante em pacientes de transplante renal e cardíaco. No período precoce do transplante hepático, pacientes que recebem uma dose de 1,5 g oral ou intravenosa de MMF tiveram níveis de MPA similares aos dos receptores de transplante renal que recebem 1 g oral ou intravenoso de MMF.
Absorção
Após a administração oral, micofenolato de mofetila sofre rápida e extensa absorção, sendo completamente metabolizado para MPA, seu metabólito ativo. A biodisponibilidade média de micofenolato de mofetila oral, baseada na AUC do MPA, está relacionada em 94% à de micofenolato de mofetila IV.
O micofenolato de mofetila pode ser mensurado, sistemicamente, durante a infusão intravenosa; entretanto, após a administração oral, micofenolato de mofetila está abaixo do limite de quantificação (0,4 mcg/mL).
No período de pós-transplante recente (< 40 dias), os pacientes de transplante renal, cardíaco e hepático apresentaram redução média da AUC do MPA de aproximadamente 30% e redução da Cmáx de aproximadamente 40% comparada ao período de pós-transplante tardio (3 - 6 meses).
No índice de infusão recomendado para pacientes submetidos a transplante renal, os valores da AUC do MPA na fase pós-transplante imediata após administração de 1 g duas vezes ao dia por via intravenosa são comparáveis àqueles observados após administração oral. Em pacientes submetidos a transplante hepático, a administração de 1 g duas vezes ao dia por via intravenosa seguida pela administração de 1,5 g duas vezes ao dia por via oral, resultou em valores de AUC do MPA semelhantes àqueles encontrados nos pacientes submetidos a transplante renal que receberam 1g duas vezes ao dia de micofenolato de mofetila.
Equivalência de formas farmacêuticas orais
A bioequivalência de micofenolato de mofetila foi avaliada. Dois comprimidos de 500 mg mostraram-se equivalentes a quatro cápsulas de 250 mg.
Distribuição
Como resultado da recirculação entero-hepática, normalmente observa-se aumento secundário na concentração plasmática do MPA em aproximadamente 6 - 12 horas após a administração da dose. A redução da área sob a curva (AUC) do MPA em aproximadamente 40% está associada à co-administração de colestiramina (4 g três vezes ao dia), indicando que existe interrupção da recirculação entero-hepática. O MPA, em concentrações clinicamente relevantes, apresenta-se ligado em 97% à albumina plasmática.
Metabolismo
O MPA é metabolizado principalmente pela glucoronil transferase (isoforma UGT1A9) para formar o glucoronídeo fenólico do MPA inativo (MPAG) In vivo, o MPAG é convertido novamente em MPA livre através da recirculação entero-hepática. Um acil-glucoronídeo menor (AcMPAG) também é formado. AcMPAG é farmacologicamente ativo e é suspeito de ser responsável por alguns dos efeitos colaterais do MMF (diarreia, leucopenia).
Eliminação
Uma porção desprezível da droga é excretada na forma de MPA (< 1% da dose) na urina. micofenolato de mofetila marcado radioativamente, quando administrado por via oral, foi completamente recuperado, sendo 93% da dose recuperada na urina e 6% recuperada nas fezes. A maior parte (aproximadamente 87%) da dose administrada foi excretada na urina sob a forma de MPAG.
Em concentrações clínicas normais, o MPA e o MPAG não são removidos pela hemodiálise. No entanto, em concentrações altas do MPAG (> 100 mcg/mL), pequenas quantidades são removidas. Por interferirem na circulação entero-hepática da droga, os sequestrantes de ácido biliar, como a colestiramina, reduzem a AUC do MPA.
A disposição de MPA depende de vários transportadores. Polipeptídeos transportadores de ânions orgânicos (OATPs) e proteína de resistência a múltiplas drogas tipo 2 (MRP2) estão envolvidos na disposição do MPA; isoformas OATP, MRP2 e proteína resistente ao câncer da mama (BCRP) são transportadores associados à excreção biliar dos glucoronídeos. A proteína de resistência a múltiplas drogas tipo 1 (MDR1) também é capaz de transportar MPA, mas a sua contribuição parece estar restrita ao processo de absorção. Nos rins, MPA e seus metabólitos interagem potencialmente com os transportadores de ânions orgânicos renais.
Farmacocinética em situações clínicas especiais
Pacientes com grave comprometimento renal
Em estudo de dose única (seis pacientes por grupo), a média das AUCs do MPA plasmático observada em indivíduos com disfunção renal crônica grave (taxa de filtração glomerular < 25 mL/min/1,73 m2) foi de 28 - 75% maior em relação à observada em voluntários saudáveis normais ou indivíduos com menor grau de comprometimento renal. A AUC do MPAG em dose única foi de três a seis vezes maior em indivíduos com disfunção renal grave em relação aos indivíduos com disfunção renal moderada ou a indivíduos normais saudáveis, concordando com a eliminação renal conhecida do MPAG. Não se estudou o efeito de doses múltiplas de micofenolato de mofetila em pacientes com disfunção renal crônica grave.
Pacientes com retardo na função do enxerto renal
Nos pacientes com retardo da função do enxerto renal pós-transplante, a AUC0-12 média do MPA foi comparável à observada em pacientes transplantados sem problemas da função do enxerto renal pós-transplante. Deve haver um aumento transitório da fração livre e da concentração plasmática do MPA em pacientes com retardo na função do enxerto. Ajuste na dose de micofenolato de mofetila não parece ser necessário. A AUC0-12 média do MPAG plasmático foi de duas a três vezes maior que em pacientes sem retardo na função do enxerto renal pós-transplante.
Em pacientes com retardo primário na função do enxerto após o transplante renal, as concentrações plasmáticas do MPAG acumularam; acúmulo do MPA, se houve, foi muito pequeno.
Pacientes com comprometimento hepático
A farmacocinética do MPAG e do MPA não foi afetada, em termos relativos, por doença do parênquima hepático em voluntários com cirrose alcoólica tratados com micofenolato de mofetila oral ou intravenoso. Os efeitos da doença hepática sobre esses processos dependem, provavelmente, da doença específica. Todavia, a doença hepática com dano predominantemente biliar, como a cirrose biliar primária, pode apresentar efeito diferente.
Idosos (>65 anos)
O comportamento de micofenolato de mofetila em idosos não foi avaliado formalmente.
Dados de segurança pré-clínicos
Em modelos experimentais, micofenolato de mofetila não foi tumorigênico. A dose mais alta testada em estudos de carcinogenicidade em animais resultou em, aproximadamente, duas a três vezes a exposição sistêmica (AUC ou Cmáx) observada em pacientes de transplante renal na dose clínica recomendada de 2 g/dia e 1,3 - 2 vezes a exposição sistêmica (AUC ou Cmáx) observada em pacientes de transplante cardíaco na dose clínica recomendada de 3 g/dia. Dois ensaios genotóxicos (ensaio de linfoma/timidina quinase em ratos e ensaio de aberração micronuclear em ratos) mostraram o potencial de micofenolato de mofetila de causar instabilidade cromossômica em doses com níveis altamente tóxicos. Outros testes genotóxicos (ensaio da mutação bacteriana, ensaio da conversão genética mitótica da levedura ou ensaio da aberração cromossômica das células ovarianas de hamster chinês) não mostraram atividade mutagênica.
O micofenolato de mofetila não apresentou efeito na fertilidade de ratos machos em doses orais de até 20 mg/kg/dia. A exposição sistêmica a essa dose representa duas a três vezes a exposição clínica na dose recomendada de 2 g/dia para pacientes de transplante renal e 1,3 - 2 vezes a exposição sistêmica em pacientes de transplante cardíaco na dose clínica recomendada de 3 g/dia. Em estudos de reprodução e fertilidade em animais fêmeas, doses orais de 4,5 mg/kg/dia causaram malformação (incluindo anoftalmia, agnatia e hidrocefalia) na primeira geração de filhotes, na ausência de toxicidade materna. A exposição sistêmica a essa dose foi aproximadamente 0,5 vez a exposição clínica na dose recomendada de 2 g/dia para pacientes de transplante renal e aproximadamente 0,3 vez a exposição clínica na dose recomendada de 3 g/dia para pacientes de transplante cardíaco. Nenhum efeito na fertilidade ou nos parâmetros reprodutivos foi observado nas fêmeas com crias ou nas gerações subsequentes.
Em estudos teratogênicos em ratos e coelhos, reabsorção fetal e malformações ocorreram em ratos na dose de 6 mg/kg/dia (incluindo anoftalmia, agnatia e hidrocefalia) e em coelhos na dose de 90 mg/kg/dia (incluindo anormalidades cardiovascular e renal, como ectopia de cordão e renal, e hérnia diafragmática e umbilical), na ausência de toxicidade materna. A exposição sistêmica a esses níveis é equivalente a cerca de menos de 0,5 vez a exposição clínica na dose recomendada de 2 g/dia para pacientes de transplante renal e aproximadamente 0,3 vez a exposição clínica na dose recomendada de 3 g/dia para pacientes de transplante cardíaco.
Os sistemas hematológico e linfático foram os primeiros a serem comprometidos em estudos toxicológicos realizados com micofenolato de mofetila em ratos, camundongos, cachorros e macacos. Esses efeitos ocorreram em níveis de exposição sistêmica que são menores que a exposição clínica na dose recomendada de 2 g/dia para pacientes de transplante renal. Efeitos gastrintestinais foram observados em cachorros em níveis de exposição sistêmica equivalentes ou menores que a exposição na dose recomendada. Efeitos gastrintestinal e renal compatíveis com desidratação também foram observados em macacos com a dose mais elevada (níveis de exposição sistêmica que são equivalentes ou maiores que a exposição clínica). O perfil de toxicidade não clínica de micofenolato de mofetila parece ser compatível com os eventos adversos observados em estudos clínicos em humanos, que atualmente fornecem dados de segurança de maior relevância para os pacientes.
Cuidados de Armazenamento
Conservar em temperatura ambiente (entre 15 e 30ºC), proteger da umidade e da luz.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Myfortic 180 mg
Um comprimido verde redondo, com bordas chanfradas e com a letra C impressa em uma face do comprimido.
Myfortic 360 mg
Um comprimido vermelho alaranjado pálido, ovaloide, com bordas chanfradas e com as letras CT impressas em uma face do comprimido.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Mensagens de Alerta
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS – 1.0068.0897
Farm. Resp.: Flavia Regina Pegorer – CRF-SP 18.150
Importado por:
Novartis Biociências S.A.
Av. Prof. Vicente Rao, 90
São Paulo - SP
CNPJ: 56.994.502/0001-30
Indústria Brasileira
Fabricado por:
Novartis Pharma Stein AG, Stein, Suíça.
Embalado por: Novartis Pharma Stein AG, Stein, Suíça ou Novartis Pharma Produktions GmbH, Wehr, Alemanha
Venda sob prescrição médica.
Esta bula foi aprovada pela ANVISA em 05/02/2016